LYTGOBI (FUTIBATINIB) tablet
1 INDICATIONS AND USAGE
LYTGOBI is indicated for the treatment of adult patients with previously treated, unresectable, locally advanced or metastatic intrahepatic cholangiocarcinoma harboring fibroblast growth factor receptor 2 (FGFR2) gene fusions or other rearrangements [see Dosage and Administration (2.1)].
This indication is approved under accelerated approval based on overall response rate and duration of response [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
LYTGOBI is a kinase inhibitor indicated for the treatment of adult patients with previously treated, unresectable, locally advanced or metastatic intrahepatic cholangiocarcinoma harboring fibroblast growth factor receptor 2 (FGFR2) gene fusions or other rearrangements. (1, 2.1)
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s). (1)
2 DOSAGE AND ADMINISTRATION
- Confirm the presence of an FGFR2 gene fusion or other rearrangement prior to initiation of treatment with LYTGOBI. (2.1)
- Recommended dose is 20 mg orally (five 4 mg tablets or one 16 mg tablet and one 4 mg tablet) once daily until disease progression or unacceptable toxicity occurs. (2.2)
- Swallow tablet whole, with or without food. (2.2)
2.1 Patient Selection
Select patients for the treatment of unresectable, locally advanced or metastatic intrahepatic cholangiocarcinoma with LYTGOBI based on the presence of an FGFR2 gene fusion or rearrangement [see Clinical Studies (14.1)]. An FDA-approved test for detection of FGFR2 gene fusions or other rearrangements in patients with unresectable, locally advanced, or metastatic intrahepatic cholangiocarcinoma for selecting patients for treatment with LYTGOBI is not available.
2.2 Recommended Dosage
The recommended dosage of LYTGOBI is 20 mg (five 4 mg tablets or one 16 mg and one 4 mg tablet) taken orally once daily until disease progression or unacceptable toxicity occurs.
Take LYTGOBI with or without food at approximately the same time each day [see Clinical Pharmacology (12.3)].
Swallow tablets whole. Do not crush, chew, split, or dissolve tablets.
If the patient misses a dose of LYTGOBI for more than 12 hours or if vomiting occurs, resume dosing with the next scheduled dose.
3 DOSAGE FORMS AND STRENGTHS
Tablets:
4 mg, round, white, film-coated tablets debossed with “4MG” on one side, and “FBN” on the other side.
16 mg, round, white, film-coated tablets debossed with “16MG” on one side and “FBN” on the other side.
Tablets: 4 mg and 16 mg (3)
5 WARNINGS AND PRECAUTIONS
- Ocular Toxicity: LYTGOBI can cause retinal pigment epithelial detachment (RPED). Perform a comprehensive ophthalmological examination including optical coherence tomography (OCT) prior to initiation of therapy, every 2 months for the first 6 months, and every 3 months thereafter and urgently at any time for visual symptoms. (2.3, 5.1)
- Hyperphosphatemia and Soft Tissue Mineralization: Increases in phosphate levels can cause hyperphosphatemia leading to soft tissue mineralization, calcinosis, nonuremic calciphylaxis and vascular calcification. Monitor for hyperphosphatemia and withhold, reduce the dose, or permanently discontinue based on duration and severity of hyperphosphatemia. (2.3, 5.2)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of reproductive potential of the potential risk to the fetus and to use effective contraception. (5.3, 8.1, 8.3)
5.1 Ocular Toxicity
Retinal Pigment Epithelial Detachment (RPED)
LYTGOBI can cause RPED, which may cause symptoms such as blurred vision.
Among 318 patients who received LYTGOBI across clinical trials [see Adverse Reactions (6.1)] where ophthalmologic monitoring did not routinely include optical coherence tomography (OCT), RPED occurred in 9% of patients. The median time to first onset of RPED was 40 days. RPED led to dose interruption of LYTGOBI in 1.3% of patients, dose reduction in 1.6% of patients, and permanent discontinuation in 0.3% of patients.
Perform a comprehensive ophthalmological examination, including OCT of the macula, prior to initiation of therapy, every 2 months for the first 6 months, and every 3 months thereafter. For onset of visual symptoms, refer patients for ophthalmologic evaluation urgently, with follow-up every 3 weeks until resolution or discontinuation of LYTGOBI.
Withhold or reduce the dose of LYTGOBI as recommended [see Dosage and Administration (2.3)].
Dry Eye/Corneal Keratitis
Among 318 patients who received LYTGOBI across clinical trials [see Adverse Reactions (6.1)], dry eye occurred in 15% of patients. Treat patients with ocular demulcents as needed.
5.2 Hyperphosphatemia and Soft Tissue Mineralization
LYTGOBI can cause hyperphosphatemia leading to soft tissue mineralization, calcinosis, nonuremic calciphylaxis, and vascular calcification. Increases in phosphate levels are a pharmacodynamic effect of LYTGOBI [see Clinical Pharmacology (12.2)]. Among 318 patients who received LYTGOBI across clinical trials [see Adverse Reactions (6.1)], hyperphosphatemia was reported in 88% of patients based on laboratory values above the upper limit of normal. The median time to onset of hyperphosphatemia was 5 days (range 3-117). Phosphate binders were received by 77% of patients who received LYTGOBI.
Monitor for hyperphosphatemia throughout treatment. Initiate a low phosphate diet and phosphate lowering therapy when serum phosphate level is ≥5.5 mg/dL. For serum phosphate levels >7 mg/dL, initiate or intensify phosphate lowering therapy and dose reduce, withhold, or permanently discontinue LYTGOBI based on duration and severity of hyperphosphatemia [see Dosage and Administration (2.3)].
5.3 Embryo-Fetal Toxicity
Based on findings in an animal study and its mechanism of action, LYTGOBI can cause fetal harm when administered to a pregnant woman. Oral administration of futibatinib to pregnant rats during the period of organogenesis caused fetal malformations, fetal growth retardation, and embryo-fetal death at maternal exposures lower than the human exposure at the clinical dose of 20 mg based on area under the curve (AUC).
Advise pregnant women of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during treatment with LYTGOBI and for 1 week after the last dose of LYTGOBI. Advise males with female partners of reproductive potential to use effective contraception during treatment with LYTGOBI and for 1 week after the last dose [see Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
The following adverse reactions are discussed elsewhere in the labeling:
- Ocular Toxicity [see Warnings and Precautions (5.1)]
- Hyperphosphatemia and Soft Tissue Mineralization [see Warnings and Precautions (5.2)]
- Most common (≥20%) adverse reactions were nail toxicity, musculoskeletal pain, constipation, diarrhea, fatigue, dry mouth, alopecia, stomatitis, abdominal pain, dry skin, arthralgia, dysgeusia, dry eye, nausea, decreased appetite, urinary tract infection, palmar-plantar erythrodysesthesia syndrome, and vomiting. (6.1)
- Most common laboratory abnormalities (≥20%) were increased phosphate, increased creatinine, decreased hemoglobin, increased glucose, increased calcium, decreased sodium, decreased phosphate, increased alanine aminotransferase, increased alkaline phosphatase, decreased lymphocytes, increased aspartate aminotransferase, decreased platelets, increased activated partial thromboplastin time, decreased leukocytes, decreased albumin, decreased neutrophils, increased creatine kinase, increased bilirubin, decreased glucose, increased prothrombin international normalized ratio, and decreased potassium. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Taiho Oncology Inc. at 1-844-878-2446 or FDA at 1-800-FDA-1088 or http://www.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflect exposure to LYTGOBI as a single agent at 20 mg orally once daily in 318 patients including 145 patients with cholangiocarcinoma and 173 patients with other advanced solid tumors. Among 318 patients who received LYTGOBI, 37% were exposed for 6 months or longer and 13% were exposed for greater than 12 months.
Previously Treated, Unresectable Locally Advanced or Metastatic Intrahepatic Cholangiocarcinoma
The safety of LYTGOBI was evaluated in Study TAS-120-101, which included 103 patients with previously treated, unresectable locally advanced or metastatic intrahepatic cholangiocarcinoma harboring FGFR2 fusions or other gene rearrangements [see Clinical Studies (14.1)]. Patients were treated with LYTGOBI 20 mg orally once daily until disease progression or unacceptable toxicity. The median duration of treatment was 9 months (range: 0.5 - 25 months).
Serious adverse reactions occurred in 39% of patients receiving LYTGOBI. Serious adverse reactions in ≥2% of patients who received LYTGOBI included pyrexia (3.9%), gastrointestinal hemorrhage (3.9%), ascites (2.9%), musculoskeletal pain (2.9%), and bile duct obstruction (2.9%).
Permanent discontinuation due to an adverse reaction occurred in 4.9% of patients who received LYTGOBI. Adverse reactions requiring permanent discontinuation of LYTGOBI in one patient each were esophagitis, oral dysesthesia, bile duct obstruction, dizziness, and anemia.
Dosage interruptions due to an adverse reaction occurred in 66% of patients who received LYTGOBI. Adverse reactions requiring dosage interruption in ≥5% of patients included hyperphosphatemia, palmar-plantar erythrodysesthesia syndrome, increased alanine aminotransferase, increased aspartate aminotransferase, and fatigue.
Dose reductions due to an adverse reaction occurred in 58% of patients who received LYTGOBI. Adverse reactions requiring dosage reductions in ≥2% of patients who received LYTGOBI included hyperphosphatemia, palmar-plantar erythrodysesthesia syndrome, fatigue, increased alanine aminotransferase, increased aspartate aminotransferase, nail toxicity, and stomatitis.
The most common (≥20%) adverse reactions were nail toxicity, musculoskeletal pain, constipation, diarrhea, fatigue, dry mouth, alopecia, stomatitis, abdominal pain, dry skin, arthralgia, dysgeusia, dry eye, nausea, decreased appetite, urinary tract infection, palmar-plantar erythrodysesthesia syndrome, and vomiting.
The most common laboratory abnormalities (≥20%) were increased phosphate, increased creatinine, decreased hemoglobin, increased glucose, increased calcium, decreased sodium, decreased phosphate, increased alanine aminotransferase, increased alkaline phosphatase, decreased lymphocyte, increased aspartate aminotransferase, decreased platelets, increased activated partial thromboplastin time, decreased leukocytes, decreased albumin, decreased neutrophils, increased creatine kinase, increased bilirubin, decreased glucose, increased prothrombin international normalized ratio, and decreased potassium.
Table 3 summarizes the adverse reactions in TAS-120-101. Table 4 summarizes laboratory abnormalities in TAS-120-101.
Clinically relevant adverse reactions occurring in ≤15% of patients included retinal pigment epithelial detachment (RPED, 7.8%).
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on LYTGOBI
Strong CYP3A Inhibitors
Avoid concomitant use of strong CYP3A inhibitors with LYTGOBI.
Futibatinib is a CYP3A substrate [see Clinical Pharmacology (12.3)]. Concomitant use of a strong CYP3A inhibitor increases futibatinib exposure [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions.
Strong CYP3A Inducers
Avoid concomitant use of strong CYP3A inducers with LYTGOBI.
Futibatinib is a CYP3A substrate [see Clinical Pharmacology (12.3)]. Concomitant use of a strong CYP3A inducer decreases futibatinib exposure [see Clinical Pharmacology (12.3)], which may reduce the effectiveness of LYTGOBI.
8 USE IN SPECIFIC POPULATIONS
- Lactation: Advise not to breastfeed. (8.2)
8.1 Pregnancy
Risk Summary
Based on findings in an animal study and its mechanism of action, LYTGOBI can cause fetal harm or loss of pregnancy when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on the use of LYTGOBI in pregnant women. Oral administration of futibatinib to pregnant rats during the period of organogenesis at maternal plasma exposures below the human exposure at the clinical dose of 20 mg resulted in fetal malformations, fetal growth retardation, and embryo-fetal death (see Data ). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Once daily oral administration of futibatinib to pregnant rats during the period of organogenesis resulted in 100% embryofetal mortality due to post-implantation loss at doses ≥10 mg/kg (approximately 5 times the recommended clinical dose of 20 mg based on body surface area, BSA). Fetal survival was unaffected at 0.5 mg/kg per day; however, once daily oral administration of futibatinib at the 0.5 mg/kg dose level (approximately 0.2 times the recommended clinical dose of 20 mg based on BSA) resulted in reduced mean fetal body weight and an increase in fetal skeletal and visceral malformations, major blood vessel variations, and reduced ossification.
8.2 Lactation
Risk Summary
There are no data on the presence of futibatinib or its metabolites in human milk or their effects on either the breastfed child or on milk production. Because of the potential for serious adverse reactions from LYTGOBI in breastfed children, advise women not to breastfeed during treatment and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
LYTGOBI can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status of females of reproductive potential prior to initiating LYTGOBI [see Use in Specific Populations (8.1)].
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with LYTGOBI and for 1 week after the last dose.
Males
Advise males with female partners of reproductive potential or who are pregnant to use effective contraception during treatment with LYTGOBI and for 1 week after the last dose.
8.4 Pediatric Use
The safety and effectiveness of LYTGOBI have not been established in pediatric patients.
Animal Toxicity Data
In 4- or 13-week repeat-dose toxicology studies in adult rats and dogs, findings included increased inorganic phosphorus and calcium in plasma, ectopic mineralization in various organs and tissues, and lesions in bone/cartilage at futibatinib exposures lower than the human exposure at the clinical dose of 20 mg. Findings in rats also included corneal lesions. Evidence of recovery in rats and dogs was observed four weeks after cessation of dosing except for the ectopic mineralization.
8.5 Geriatric Use
Of the 103 patients treated with LYTGOBI in Study TAS-120-101, 22% were 65 years or older. Based on available data, no overall differences in safety or effectiveness of LYTGOBI have been observed between patients 65 years of age and older and younger adult patients.
8.7 Hepatic Impairment
There are limited data in patients treated with LYTGOBI with total bilirubin >1.5🞨 ULN, and patients with cholangiocarcinoma with total bilirubin >3🞨 ULN were excluded from enrollment in clinical trials. Patients with cirrhosis and total bilirubin >1.5🞨 ULN to 3🞨 ULN may have the potential for increased adverse reactions compared to patients with normal hepatic function due to higher unbound futibatinib exposure [see Clinical Pharmacology (12.3)].
11 DESCRIPTION
Futibatinib is a kinase inhibitor with the chemical name 1-[(3S)-3-{4-amino-3-[(3,5-dimethoxyphenyl)ethynyl]- 1H-pyrazolo[3,4-d]pyrimidin-1-yl}pyrrolidin-1-yl]prop-2-en-1-one. Futibatinib has a molecular formula of C22H22N6O3 and molecular mass of 418.45 g/mole. Futibatinib has the following chemical structure:

Futibatinib is a white crystalline powder. The solubility of futibatinib is pH dependent with decreasing solubility with increasing pH, being practically insoluble at pH 3 or higher. Futibatinib is insoluble in water and poorly soluble in common solvents. LYTGOBI is supplied as 4 mg and 16 mg film-coated tablets for oral administration. Each tablet contains inactive ingredients of corn starch, crospovidone, hydroxypropyl cellulose, lactose monohydrate, magnesium stearate, mannitol, microcrystalline cellulose, and sodium lauryl sulfate. The film coating material contains hypromellose, magnesium stearate, polyethylene glycol, and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Futibatinib is a small molecule kinase inhibitor of FGFR 1, 2, 3, and 4 with IC50 values of less than 4 nM. Futibatinib covalently binds FGFR. Constitutive FGFR signaling can support the proliferation and survival of malignant cells. Futibatinib inhibited FGFR phosphorylation and downstream signaling and decreased cell viability in cancer cell lines with FGFR alterations including FGFR fusions/rearrangements, amplifications, and mutations. Futibatinib demonstrated anti-tumor activity in mouse and rat xenograft models of human tumors with activating FGFR genetic alterations.
12.2 Pharmacodynamics
Serum Phosphate
Futibatinib increased serum phosphate levels due to FGFR inhibition. Serum phosphate increased with increasing futibatinib exposure across the dose range of 4 to 24 mg orally once daily (0.2 to 1.2 times the recommended dose), with increased risk of hyperphosphatemia at higher futibatinib exposure.
Cardiac Electrophysiology
At four times the approved recommended dose, LYTGOBI does not cause clinically significant QTc interval prolongation.
12.3 Pharmacokinetics
The pharmacokinetics of futibatinib were observed at steady state in patients with advanced solid tumors unless otherwise specified at the approved recommended dosage and are presented as mean (%CV) unless otherwise specified.
Futibatinib maximum concentration (Cmax) is 144 mg/mL (50%) and systemic exposure (AUC) is 790 ng∙hr/mL (45%). Futibatinib exposure (AUC) increased in dose proportional manner over the dose range from 4 to 24 mg orally once daily (0.2 to 1.2 times the approved recommended dosage). No accumulation is observed at steady state.
Absorption
Futibatinib median (min, max) time to maximum plasma concentration (Tmax) is 2 hours (1.2, 23 hours).
Effect of Food
Futibatinib Cmax decreased by 42% with no clinically significant differences in AUC in healthy subjects following administration of LYTGOBI with a high-fat and high-calorie meal (900 to 1000 calories with approximately 50% from fat).
Distribution
The apparent (oral) volume of distribution is 66 L (18%). Futibatinib plasma protein binding is 95%, primarily to albumin and α1-acid glycoprotein.
Elimination
Futibatinib elimination half-life is 2.9 hours (27%) with an apparent (oral) clearance of 20 L/hr (23%).
Metabolism
Futibatinib is primarily metabolized by CYP3A and to a lesser extent by CYP2C9 and CYP2D6. Unchanged futibatinib accounts for 59% of radioactivity in plasma in healthy subjects.
Excretion
Following a single oral dose of radiolabeled futibatinib 20 mg, approximately 91% of the dose was recovered in feces and 9% in urine.
Specific Populations
No clinically significant differences in the pharmacokinetics of futibatinib were observed based on age (18 to 82 years), sex, race (White, Asian, and African American), body weight (36 to 152 kg), or creatinine clearance (CLcr) 30 to 89 mL/min (estimated by Cockcroft-Gault). The effect of CLcr of 15 to 29 mL/min or renal dialysis in end-stage renal disease (CLcr <15 mL/min) on futibatinib pharmacokinetics is unknown.
Patients with Hepatic Impairment
No clinically significant differences in the pharmacokinetics of futibatinib were observed in subjects with mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN or total bilirubin >1 to 1.5🞨 ULN and any AST or Child-Pugh A) or moderate to severe hepatic impairment (Child-Pugh B to C) with total bilirubin < 1.5 🞨 ULN. The effect of moderate to severe hepatic impairment without cirrhosis on futibatinib pharmacokinetics is unknown.
In subjects with cirrhosis and total bilirubin >1 to 1.5🞨 ULN, mean unbound futibatinib AUC and Cmax increased by 1.7-fold relative to subjects with normal hepatic function. In subjects with cirrhosis and total bilirubin >1.5🞨 ULN, mean unbound futibatinib AUC and Cmax increased by 3.2-fold and 2.4-fold, respectively, compared to values in healthy subjects.
Drug Interaction Studies
Clinical Studies
Strong CYP3A inhibitors: Futibatinib Cmax increased by 1.5-fold and AUC by 1.4-fold following concomitant use of multiple doses of itraconazole (strong CYP3A inhibitor).
Strong CYP3A inducers: Futibatinib Cmax decreased by 53% and AUC by 64% following concomitant use of multiple doses of rifampin (strong CYP3A inducer).
Other Drugs: No clinically significant differences in futibatinib pharmacokinetics were observed with concomitant use of multiple doses of quinidine (P-gp inhibitor) or lansoprazole (proton pump inhibitor).
No clinically significant differences in the pharmacokinetics of the following drugs were observed following concomitant use of multiple doses of LYTGOBI: midazolam (CYP3A substrate), digoxin (P-pg substrate), and rosuvastatin (BCRP substrate).
In Vitro Studies
CYP Enzymes: Futibatinib does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6 and does not induce CYP1A2 or CYP2B6.
Transporter Systems: Futibatinib is not a substrate of OATP1B1 or OATP1B3. Futibatinib does not inhibit OAT1, OAT3, OCT2, OATP1B1, OATP1B3, MATE1, or MATE2K.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with futibatinib.
Futibatinib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay. Futibatinib was clastogenic in an in vitro chromosomal aberration assay. Futibatinib was not clastogenic in a rat bone marrow micronucleus assay or a rat DNA damaging (Comet) assay.
Dedicated fertility studies with futibatinib have not been conducted.
14 CLINICAL STUDIES
14.1 Cholangiocarcinoma
TAS-120-101 (NCT02052778), a multicenter, open-label, single-arm trial, evaluated the efficacy of LYTGOBI in 103 patients with previously treated, unresectable, locally advanced or metastatic intrahepatic cholangiocarcinoma. The presence of FGFR2 fusions or other rearrangements was determined in 102 enrolled patients (99%) using next generation sequencing (NGS) testing. Qualifying in-frame fusions and other rearrangements were predicted to have a breakpoint within intron 17/exon 18 of the FGFR2 gene leaving the FGFR2 kinase domain intact.
Patients received LYTGOBI at a dosage of 20 mg orally once daily until disease progression or unacceptable toxicity. The major efficacy outcome measures were overall response rate (ORR) and duration of response (DoR) as determined by an independent review committee (IRC) according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1.
The trial population characteristics were: Median age was 58 years (range: 22 to 79 years) with 22% of patients ≥65 years, 56% were female, race was: 50% White, 29% Asian, 8% Black or African American, 1% Native Hawaiian or Other Pacific Islander, 13% unknown, baseline Eastern Cooperative Oncology Group (ECOG) performance status of 0 (47%) or 1 (53%). Seventy-eight percent (78%) of patients had in-frame FGFR2 gene fusions and the most commonly identified FGFR2 fusion partner was BICC1 (n=24, 23%). Twenty-two percent (22%) of patients had other FGFR2 rearrangements that may not be in-frame with the partner gene or the partner gene was not identifiable.
All patients had received at least 1 prior systemic therapy, 30% had 2 prior lines of therapy, and 23% had 3 or more prior lines of therapy. All patients received a prior platinum-based therapy including 91% with prior gemcitabine/cisplatin.
Efficacy results are summarized in Table 5. The median time to response was 2.5 months (range 0.7 – 7.4 months).
16 HOW SUPPLIED/STORAGE AND HANDLING
LYTGOBI tablets are round, white, and film-coated. The 4 mg tablets are debossed with “4MG” on one side, and “FBN” on the other side. The 16 mg tablets are debossed with “16MG” on one side, and “FBN” on the other. Tablets are packaged in blister cards and supplied in child-resistant DosePak® as follows:
- 20 mg daily dose: Each carton contains 1 blister card containing a 7-day supply (35 tablets; 4 mg futibatinib per tablet). [NDC-64842-0120-6]
- 20 mg daily dose: Each carton contains 1 blister card containing a 7-day supply (14 tablets; 7 tablets each of 4 mg and 16 mg futibatinib per tablet). [NDC-64842-0128-8]
- 16 mg daily dose: Each carton contains 1 blister card containing a 7-day supply (28 tablets; 4 mg futibatinib per tablet). [NDC-64842-0120-5]
- 16 mg daily dose: Each carton contains 1 blister card containing a 7-day supply (7 tablets; 16 mg futibatinib per tablet). [NDC-64842-0127-7]
- 12 mg daily dose: Each carton contains 1 blister card containing a 7-day supply (21 tablets; 4 mg futibatinib per tablet). [NDC-64842-0120-4]
Store LYTGOBI tablets at room temperature 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F to 86°F). ([see USP Controlled Room Temperature]).
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Ocular Toxicity
Advise patients that LYTGOBI may cause ocular toxicity including RPED and to immediately inform their healthcare provider if they experience any visual changes [see Warnings and Precautions (5.1)]. Advise patients to use artificial tears, or hydrating or lubricating eye gels to prevent or treat dry eyes [see Warnings and Precautions (5.1)].
Hyperphosphatemia and Soft Tissue Mineralization
Inform patients that LYTGOBI may cause hyperphosphatemia and soft tissue mineralization and to immediately inform their healthcare provider of any symptoms related to acute change in phosphate levels such as muscle cramps, numbness, or tingling around the mouth [see Warnings and Precautions 5.2)].
Nail Disorders
Advise patients that LYTGOBI may cause nail disorders [see Adverse Reactions (6.1)].
Embryo-Fetal Toxicity
- Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of pregnancy [see Warnings and Precautions (5.3) and Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception while on LYTGOBI and for 1 week after the last dose [see Use in Specific Populations (8.3)].
- Advise males with female partners of reproductive potential or who are pregnant to use effective contraception during treatment and for 1 week after receiving the last dose of LYTGOBI [see Use in Specific Populations (8.3)].
Lactation
- Advise patients not to breastfeed during treatment with LYTGOBI and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Administration
- Instruct patients to not crush, chew, split or dissolve tablets.
- Instruct patients if they miss a dose by 12 or more hours or if they vomit after taking a dose, resume dosing with the next scheduled dose. Extra tablets should not be taken to make up for the missed dose [see Dosage and Administration (2.2)].
Drug Interactions
Advise patients to inform their healthcare providers of all concomitant medications, including prescription medicines, over-the-counter drugs, and herbal products. Advise patients to avoid grapefruit products during treatment with LYTGOBI [see Drug Interactions (7.1)].
Manufactured for:
Taiho Pharmaceutical Co., Ltd.
Japan
Distributed by:
Taiho Oncology, Inc.
Princeton, NJ 08540 USA
LYTGOBI is a trademark of Taiho Pharmaceutical Co., Ltd.
U.S. Patent Nos. 9,108,973, 10,434,103, and 11,833,151
© 2025 Taiho Oncology, Inc. All rights reserved
PRINCIPAL DISPLAY PANEL - Blister 4mg 35ct Card
NDC 64842-0120-6
LYTGOBI®
(futibatinib) tablets
4 mg per tablet
Rx Only
Swallow tablets whole. Do not crush, chew, split, or dissolve.
20 mg daily dose. Take five 4 mg tablets once daily.
TAIHO ONCOLOGY

PRINCIPAL DISPLAY PANEL - Carton 4mg 35ct
NDC 64842-0120-6
Rx Only
LYTGOBI®
(futibatinib) tablets
20 mg daily dose
Take five 4 mg tablets orally once daily
4 mg per tablet
One blister card
containing 35 tablets
7-day supply
TAIHO ONCOLOGY, INC.

PRINCIPAL DISPLAY PANEL - Blister 4mg 28ct Card
NDC 64842-0120-5
LYTGOBI®
(futibatinib) tablets
4 mg per tablet
Rx Only
Swallow tablets whole. Do not crush, chew, split, or dissolve.
16 mg daily dose. Take four 4 mg tablets once daily.
TAIHO ONCOLOGY

PRINCIPAL DISPLAY PANEL - Carton 4mg 28ct
NDC 64842-0120-5
Rx Only
LYTGOBI®
(futibatinib) tablets
16 mg daily dose
Take four 4 mg tablets orally once daily
4 mg per tablet
One blister card
containing 28 tablets
7-day supply
TAIHO ONCOLOGY, INC.

PRINCIPAL DISPLAY PANEL - Blister 4mg 21ct Card
NDC 64842-0120-4
LYTGOBI®
(futibatinib) tablets
4 mg per tablet
Rx Only
Swallow tablets whole. Do not crush, chew, split, or dissolve.
12 mg daily dose. Take three 4 mg tablets once daily.
TAIHO ONCOLOGY

PRINCIPAL DISPLAY PANEL - Carton 4mg 21ct
NDC 64842-0120-4
Rx Only
LYTGOBI®
(futibatinib) tablets
12 mg daily dose
Take three 4 mg tablets orally once daily
4 mg per tablet
One blister card
containing 21 tablets
7-day supply
TAIHO ONCOLOGY, INC.

PRINCIPAL DISPLAY PANEL - Blister 16mg 7ct Card
NDC 64842-0127-7
LYTGOBI®
(futibatinib) tablets
16 mg daily dose. Take one
16 mg tablet once daily.
Rx Only
Swallow tablet whole. Do not crush, chew, split, or dissolve.
TAIHO ONCOLOGY, INC.
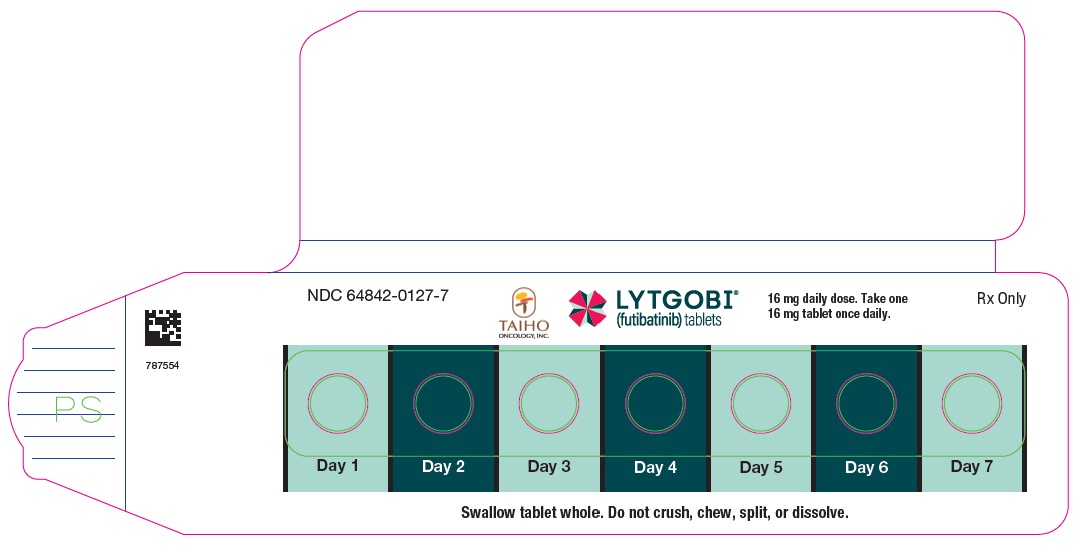
PRINCIPAL DISPLAY PANEL - Carton 16mg 7ct
NDC 64842-0127-7
Rx Only
LYTGOBI®
(futibatinib) tablets
16 mg daily dose
Take one 16 mg tablet orally once daily
16 mg per tablet
Contains: One blister
card containing 7 tablets
7-day supply (7 tablets)
TAIHO ONCOLOGY, INC.

PRINCIPAL DISPLAY PANEL - Blister 16mg 14ct Card
NDC 64842-0128-8
LYTGOBI®
(futibatinib) tablets
20 mg daily dose. Take one 4 mg tablet
and one 16 mg tablet once daily.
Rx Only
Swallow tablets whole. Do not crush, chew, split, or dissolve tablets.
TAIHO ONCOLOGY, INC.
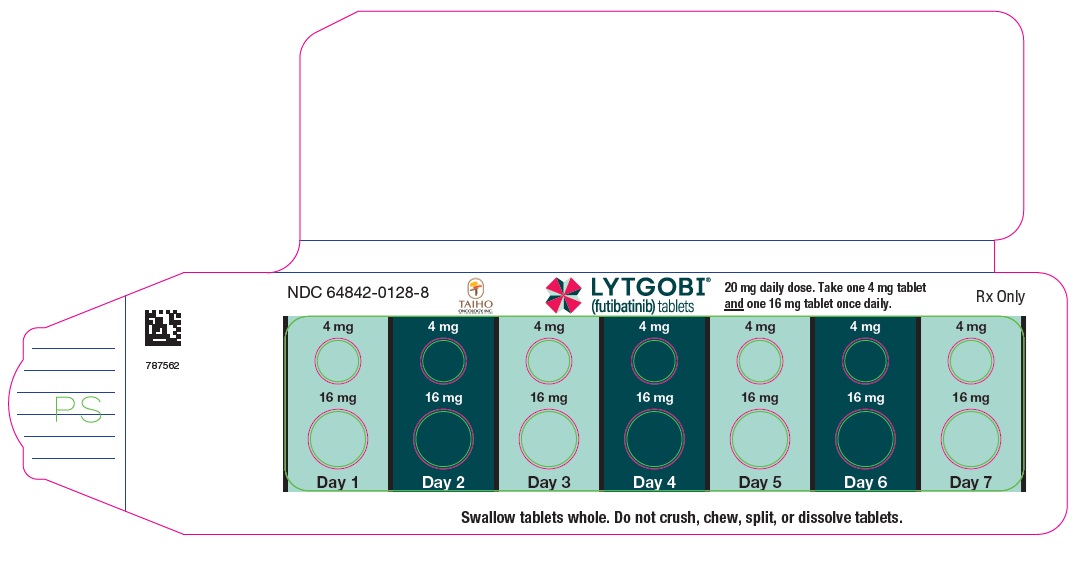
PRINCIPAL DISPLAY PANEL - Carton 16mg 14ct
NDC 64842-0128-8
Rx Only
LYTGOBI®
(futibatinib) tablets
20 mg daily dose
Take one 4 mg tablet and one
16 mg tablet orally once daily
4 mg per tablet (7 tablets)
16 mg per tablet (7 tablets)
Contains: One blister
card containing 14 tablets
7-day supply (14 tablets)
TAIHO ONCOLOGY, INC.

Guideline Central and select third party use “cookies” on this website to enhance the user experience.
This technology helps us gather statistical and analytical information to optimize the relevant content for you.
The user also has the option to opt-out which may have an effect on the browsing experience.