Kerydin (Tavaborole) solution
1 INDICATIONS AND USAGE
KERYDIN (tavaborole) topical solution, 5% is an oxaborole antifungal indicated for the treatment of onychomycosis of the toenails due to Trichophyton rubrum or Trichophyton mentagrophytes.
KERYDIN is an oxaborole antifungal indicated for the topical treatment of onychomycosis of the toenails due to Trichophyton rubrum or Trichophyton mentagrophytes. (1)
2 DOSAGE AND ADMINISTRATION
Apply KERYDIN to affected toenails once daily for 48 weeks.
KERYDIN should be applied to the entire toenail surface and under the tip of each toenail being treated.
KERYDIN is for topical use only and not for oral, ophthalmic, or intravaginal use.
3 DOSAGE FORMS AND STRENGTHS
KERYDIN topical solution, 5% is a clear, colorless alcohol-based solution. Each milliliter of solution contains 43.5 mg (5% w/w) of tavaborole.
Solution, 5%. (3)
6 ADVERSE REACTIONS
Common adverse reactions occurring in ≥1% in subjects treated with KERYDIN included application site exfoliation, ingrown toenail, application site erythema, and application site dermatitis. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer, Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In two clinical trials, 791 subjects were treated with KERYDIN. The most commonly reported adverse reactions are listed below (Table 1).
6.2 Postmarketing Experience
The following adverse reactions have been identified during postmarketing use of KERYDIN. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug product exposure:
Hypersensitivity; contact allergy
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on KERYDIN use in pregnant women to inform a drug associated risk for major birth defects, miscarriage or adverse maternal or fetal outcomes. In oral animal reproductive studies, administration of tavaborole during the period of organogenesis resulted in embryofetal toxicity and malformations at 570 times the Maximum Recommended Human Dose (MRHD) based on Area Under the Curve (AUC) comparisons in rats and embryofetal toxicity at 155 times the MRHD based on AUC comparisons in rabbits. Embryofetal toxicity was noted following dermal administration in rabbits up to 36 times the MRHD based on AUC comparisons [see Data ].
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies carry some risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects in the U.S. general population is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
Oral administration:
In an oral embryofetal development study in rats, oral doses of 30, 100, and 300 mg/kg/day tavaborole were administered during the period of organogenesis (gestational days 6–19) to pregnant female rats. In the presence of maternal toxicity, embryofetal toxicity (increased embryofetal resorption and/or deaths) and drug-related skeletal malformations and variations suggestive of delayed development (i.e., a delay in ossification) were noted in fetuses at 300 mg/kg/day tavaborole [570 times the MRHD based on AUC comparisons]. No developmental toxicity was noted in rats at 100 mg/kg/day tavaborole (26 times the MRHD based on AUC comparisons).
In an oral embryofetal development study in rabbits, oral doses of 15, 50, and 150 mg/kg/day tavaborole were administered during the period of organogenesis (gestational days 7–19) to pregnant female rabbits. In the presence of maternal toxicity, excessive embryofetal mortality due to post-implantation loss was noted at 150 mg/kg/day tavaborole. No drug related malformations were noted in rabbits at 150 mg/kg/day tavaborole (155 times the MRHD based on AUC comparisons). No embryofetal mortality was noted in rabbits at 50 mg/kg/day tavaborole (16 times the MRHD based on AUC comparisons).
In an oral pre- and post-natal development study in rats, oral doses of 15, 60, and 100 mg/kg/day tavaborole were administered from the beginning of organogenesis (gestation day 6) through the end of lactation (lactation day 20). In the presence of minimal maternal toxicity, no embryofetal toxicity or effects on postnatal development were noted at 100 mg/kg/day (29 times the MRHD based on AUC comparisons).
Topical administration:
In a dermal embryofetal development study in rabbits, topical doses of 1%, 5%, and 10% tavaborole solution were administered during the period of organogenesis (gestational days 6–28) to pregnant female rabbits. A dose dependent increase in dermal irritation at the treatment site was noted at 5% and 10% tavaborole solution. A decrease in fetal bodyweight was noted at 10% tavaborole solution. No drug related malformations were noted in rabbits at 10% tavaborole solution (36 times the MRHD based on AUC comparisons). No embryofetal toxicity was noted in rabbits at 5% tavaborole solution (26 times the MRHD based on AUC comparisons).
8.2 Lactation
Risk Summary
There is no information available on the presence of KERYDIN in human milk, the effects of the drug on the breastfed infant or the effects of the drug on milk production after topical application of KERYDIN to women who are breastfeeding. KERYDIN is systemically absorbed. The lack of clinical data during lactation precludes a clear determination of the risk of KERYDIN to a breastfed infant. Therefore, the developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for KERYDIN and any potential adverse effects on the breastfed child from KERYDIN or from the underlying maternal condition.
8.4 Pediatric Use
The safety and efficacy of KERYDIN were established in patients 6 years of age and older. Use of KERYDIN in these age groups is supported by evidence from adequate and well-controlled studies of KERYDIN in adults with additional data from an open-label pharmacokinetics study of tavaborole in subjects 12 years to less than 17 years old [see Clinical Pharmacology (12.3)].
8.5 Geriatric Use
In clinical trials of 791 subjects who were exposed to KERYDIN, 19% were 65 years of age and over, while 4% were 75 years of age and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
11 DESCRIPTION
KERYDIN (tavaborole) topical solution, 5% contains tavaborole, 5% (w/w) in a clear, colorless alcohol-based solution for topical use. The active ingredient, tavaborole, is an oxaborole antifungal with the chemical name of 5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole. The chemical formula is C7H6BFO2, the molecular weight is 151.93 and the structural formula is:
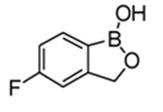
Tavaborole is a white to off-white powder. It is slightly soluble in water and freely soluble in ethanol and propylene glycol.
Each mL of KERYDIN contains 43.5 mg of tavaborole. Inactive ingredients include alcohol, edetate calcium disodium, and propylene glycol.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
KERYDIN is an oxaborole antifungal [see Clinical Pharmacology (12.4) ].
12.2 Pharmacodynamics
At therapeutic doses, KERYDIN is not expected to prolong QTc to any clinically relevant extent.
12.3 Pharmacokinetics
Tavaborole undergoes extensive metabolism. Renal excretion is the major route of elimination of the metabolites.
In a clinical pharmacology trial of six healthy adult male volunteers who received a single topical application of 5% 14C-tavaborole solution, tavaborole conjugates and metabolites were shown to be excreted primarily in the urine.
The pharmacokinetics (PK) of tavaborole was investigated in 24 adult subjects with distal subungual onychomycosis involving at least 4 toenails (including at least 1 great toenail) following a single dose and a 2-week daily topical application of 200 μL of a 5% solution of tavaborole to all ten toenails and 2 mm of skin surrounding each toenail. Steady state was achieved after 14 days of dosing. After a single dose, the mean (± standard deviation) peak concentration (Cmax) of tavaborole was 3.5 ± 2.3 ng/mL (n=21 with measurable concentrations, range 0.618–10.2 ng/mL, LLOQ=0.5 ng/mL), and the mean AUClast ± SD was 44.4 ± 25.5 ng*hr/mL (n=21). After 2 weeks of daily dosing, the mean Cmax ± SD was 5.2 ± 3.5 ng/mL (n=24, range 1.5–12.8 ng/mL), and the mean AUCτ ± SD was 75.8 ± 44.5 ng*hr/mL.
In another study PK of tavaborole was investigated in 22 subjects aged 12 years to less than 17 years with distal subungual onychomycosis involving at least 4 toenails (including at least 1 great toenail with at least 20% involvement) following once daily application of 5% solution of tavaborole to all ten toenails and 2 mm of skin surrounding each toenail for 29 days. On Day 29, the mean ± SD Cmax was 5.9 ± 4.9 ng/mL (n=21 with measurable concentrations, range 1.0 –16.4 ng/mL, LLOQ=0.5 ng/mL), and the mean ± SD AUC0-24 was 76.0 ± 62.5 ng*hr/mL.
Drug Interaction Studies
In Vitro Studies
In vitro studies have shown that tavaborole, at therapeutic concentrations, neither inhibits nor induces cytochrome P450 (CYP450) enzymes.
12.4 Microbiology
Mechanism of Action
The mechanism of action of tavaborole is inhibition of fungal protein synthesis. Tavaborole inhibits protein synthesis by inhibition of an aminoacyl-transfer ribonucleic acid (tRNA) synthetase (AARS).
Activity in vitro and in clinical infections
Tavaborole has been shown to be active against most strains of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1) ]:
Trichophyton rubrum
Trichophyton mentagrophytes
Mechanism of Resistance
Trichophyton mentagrophytes and Trichophyton rubrum strains from isolates collected in the clinical trials have not demonstrated resistance following repeated exposure to tavaborole.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In an oral carcinogenicity study in Sprague-Dawley rats, oral doses of 12.5, 25, and 50 mg/kg/day tavaborole were administered to rats once daily for 104 weeks. No drug related neoplastic findings were noted at oral doses up to 50 mg/kg/day tavaborole (14 times the MRHD based on AUC comparisons).
In a dermal carcinogenicity study in CD-1 mice, topical doses of 5%, 10%, and 15% tavaborole solution were administered to mice once daily for 104 weeks. No drug related neoplastic findings were noted at topical doses up to 15% tavaborole solution (89 times the MRHD based on AUC comparisons).
Tavaborole revealed no evidence of mutagenic or clastogenic potential based on the results of two in vitro genotoxicity tests (Ames assay and Human lymphocyte chromosomal aberration assay) and one in vivo genotoxicity test (rat micronucleus assay).
No effects on fertility were observed in male and female rats that were administered oral doses up to 300 mg/kg/day tavaborole (107 times the MRHD based on AUC comparisons) prior to and during early pregnancy.
14 CLINICAL STUDIES
The efficacy and safety of KERYDIN was evaluated in two multicenter, double-blind, randomized, vehicle-controlled trials. KERYDIN or vehicle was applied once daily for 48 weeks in subjects with 20% to 60% clinical involvement of the target toenail, without dermatophytomas or lunula (matrix) involvement.
A total of 1194 subjects (795 KERYDIN, 399 Vehicle) 18 to 88 years of age, 82% male, 84% white, participated in these two trials. Efficacy assessments were made at 52 weeks following a 48-week treatment period.
The Complete Cure efficacy endpoint included negative mycology (negative KOH wet mount and negative fungal culture) and Completely Clear Nail (no clinical evidence of onychomycosis as evidenced by a normal toenail plate, no onycholysis, and no subungual hyperkeratosis). Efficacy results from the two trials are summarized in Table 2.
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
KERYDIN (tavaborole) topical solution, 5% is a clear, colorless solution supplied in an amber glass bottle with a screw cap. At initial use, the screw cap is replaced with the dropper assembly.
KERYDIN (tavaborole) topical solution, 5% is supplied in the following presentations:
NDC 10337-905-10: One bottle containing 10 mL of solution with one glass pointed-tip dropper.
NDC 10337-905-44: One bottle containing 4 mL of solution with one glass pointed-tip dropper.
16.2 Storage and Handling
Store at 20–25°C (68–77°F); excursions permitted to 15–30°C (59–86°F) [see USP Controlled Room Temperature].
CAUTION: Flammable. Keep away from heat and flame.
Discard product within 3 months after insertion of the dropper.
Keep bottle tightly closed. Keep out of reach of children.
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Patient Information and Instructions for Use)
The patient should be told the following:
-
The impact of nail polish or other cosmetic nail products on the efficacy of KERYDIN has not been evaluated. -
Inform a health care professional if the area of application shows signs of persistent irritation (for example, redness, itching, swelling). -
Product is flammable. Avoid use near heat or open flame.
Manufactured for:
Pfizer Labs, Division of Pfizer Inc, NY, NY 10017
Distributed by:
PharmaDerm®
A division of Fougera Pharmaceuticals Inc.
Melville, New York 11747 USA
KERYDIN® is a trademark of Anacor Pharmaceuticals, Inc.
© 2015 Anacor Pharmaceuticals, Inc.
U.S. Patent Nos. 7,767,657 and 7,582,621
LAB-1202-2.0
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised August/2018
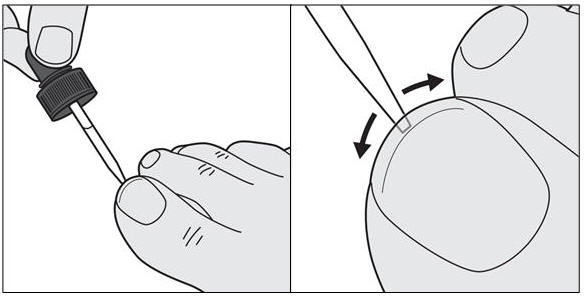
Instructions for Use
KERYDIN® (ker' i din)
(tavaborole) topical solution, 5%
Read the Instructions for Use that comes with KERYDIN before you start using it. Talk to your healthcare provider if you have any questions.
How to apply KERYDIN:
Your toenails should be clean and dry before you apply KERYDIN.
This Patient Information and Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured for: Pfizer Labs, Division of Pfizer Inc, NY, NY 10017
Distributed by: PharmaDerm®, a division of Fougera Pharmaceuticals, Inc., Melville, New York 11747 USA
LAB-1204-2.0
Revised: 08/2018
PRINCIPAL DISPLAY PANEL - 10 mL Bottle Label
PharmaDerm®
NDC 10337-905-10
Kerydin®
(TAVABOROLE)
TOPICAL SOLUTION, 5%
For Topical Use Only
10 mL
Not for oral, ophthalmic, or intravaginal use
Rx only

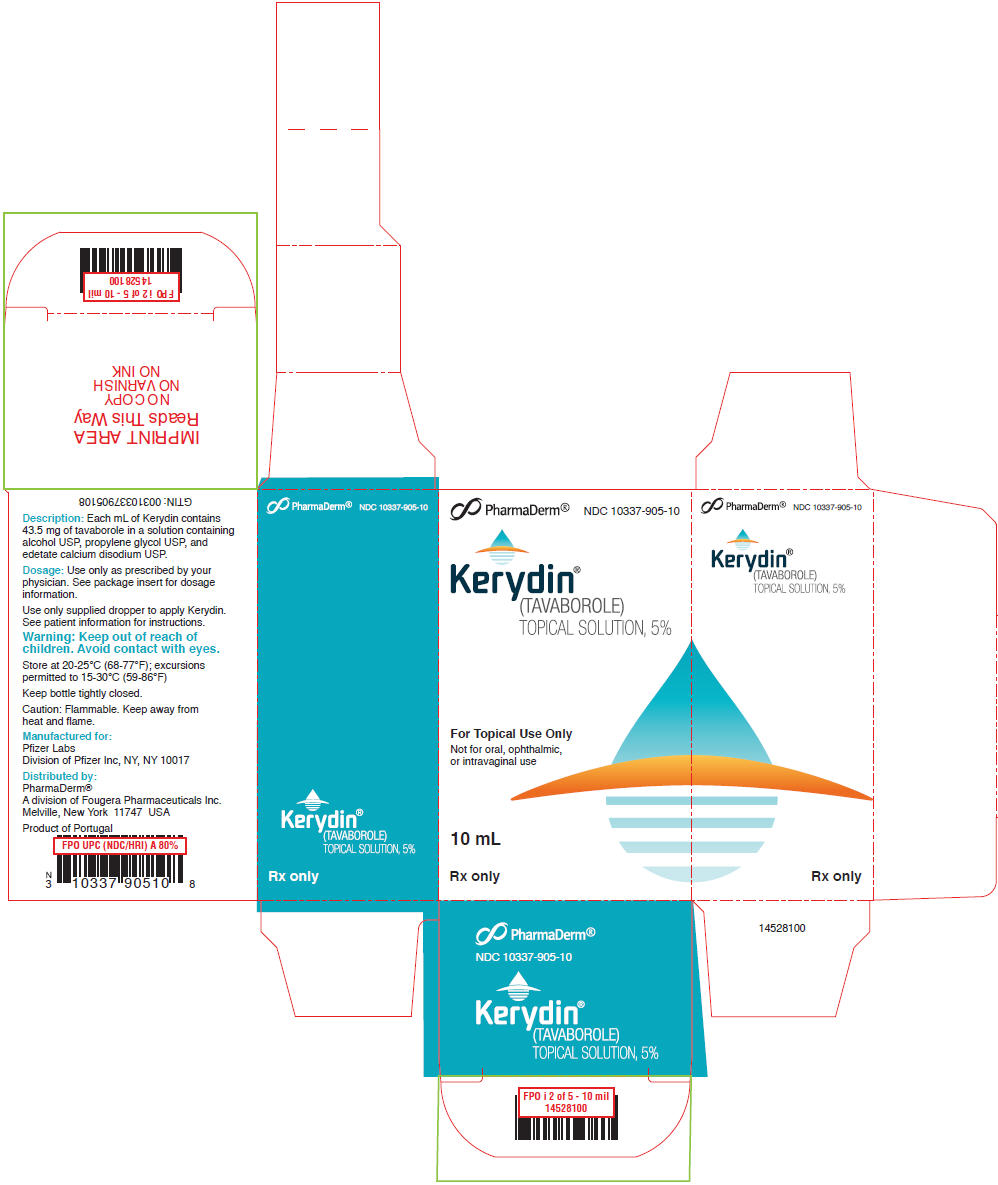
PRINCIPAL DISPLAY PANEL - 10 mL Bottle Carton
PharmaDerm®
NDC 10337-905-10
Kerydin®
(TAVABOROLE)
TOPICAL SOLUTION, 5%
For Topical Use Only
Not for oral, ophthalmic,
or intravaginal use
10 mL
Rx only
Guideline Central and select third party use “cookies” on this website to enhance the user experience.
This technology helps us gather statistical and analytical information to optimize the relevant content for you.
The user also has the option to opt-out which may have an effect on the browsing experience.