TREMFYA (Guselkumab) injection
1 INDICATIONS AND USAGE
TREMFYA is an interleukin-23 antagonist indicated for the treatment of:
- adults and pediatric patients 6 years of age and older who also weigh at least 40 kg with moderate-to-severe plaque psoriasis and who are candidates for systemic therapy or phototherapy. (1.1)
- adults and pediatric patients 6 years of age and older who also weigh at least 40 kg with active psoriatic arthritis. (1.2)
- adults with moderately to severely active ulcerative colitis. (1.3)
- adults with moderately to severely active Crohn's disease. (1.4)
1.1 Plaque Psoriasis
TREMFYA is indicated for the treatment of adults and pediatric patients 6 years of age and older who also weigh at least 40 kg with moderate-to-severe plaque psoriasis and who are candidates for systemic therapy or phototherapy.
1.2 Psoriatic Arthritis
TREMFYA is indicated for the treatment of adults and pediatric patients 6 years of age and older who also weigh at least 40 kg with active psoriatic arthritis.
1.3 Ulcerative Colitis
TREMFYA is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis.
1.4 Crohn's Disease
TREMFYA is indicated for the treatment of adult patients with moderately to severely active Crohn's disease.
2 DOSAGE AND ADMINISTRATION
- For the treatment of ulcerative colitis or Crohn’s disease: Obtain liver enzymes and bilirubin levels prior to initiating treatment with TREMFYA. (2.1, 5.4).
- For the treatment of plaque psoriasis or psoriatic arthritis, if clinically indicated, evaluate liver enzymes and bilirubin at baseline prior to initiating treatment with TREMFYA (2.1, 5.4).
- Complete all age-appropriate vaccinations as recommended by current immunization guidelines prior to treatment initiation. (2.1)
Recommended Dosage
Plaque Psoriasis
Adults
- 100 mg administered by subcutaneous injection at Week 0, Week 4, and every 8 weeks thereafter. (2.2)
Pediatric Patients 6 Years of Age and Older Who Also Weigh at Least 40 kg
- 100 mg administered by subcutaneous injection at Week 0, Week 4, and every 8 weeks thereafter. (2.2)
Psoriatic Arthritis
Adults
- 100 mg administered by subcutaneous injection at Week 0, Week 4, and every 8 weeks thereafter. TREMFYA can be used alone or in combination with a conventional DMARD (e.g., methotrexate). (2.3)
Pediatric Patients 6 Years of Age and Older Who Also Weigh at Least 40 kg
- 100 mg administered by subcutaneous injection at Week 0, Week 4, and every 8 weeks thereafter. TREMFYA may be administered alone or in combination with a conventional disease-modifying antirheumatic drug (e.g., methotrexate). (2.3)
Ulcerative Colitis and Crohn’s Disease
- Induction: 200 mg administered by intravenous infusion over at least one hour at Week 0, Week 4, and Week 8 or 400 mg administered by subcutaneous injection at Week 0, Week 4, and Week 8. (2.4)
- Maintenance: 100 mg administered by subcutaneous injection at Week 16, and every 8 weeks thereafter, or 200 mg administered by subcutaneous injection at Week 12, and every 4 weeks thereafter. Use the lowest effective recommended dosage to maintain therapeutic response. (2.4)
2.1 Recommended Evaluations and Immunizations Prior to Treatment Initiation
- Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with TREMFYA [see Warnings and Precautions (5.3)].
- For the treatment of ulcerative colitis or Crohn’s disease, obtain liver enzymes and bilirubin levels prior to initiating treatment with TREMFYA [see Warnings and Precautions (5.4)].
- For the treatment of plaque psoriasis or psoriatic arthritis, if clinically indicated, evaluate liver enzymes and bilirubin prior to initiating treatment with TREMFYA [see Warnings and Precautions (5.4)].
- Complete all age-appropriate vaccinations according to current immunization guidelines [see Warnings and Precautions (5.5)].
2.2 Recommended Dosage for Moderate-to-Severe Plaque Psoriasis
Administer TREMFYA by subcutaneous injection at Week 0, Week 4, and every 8 weeks thereafter.
Adults
The recommended dose is 100 mg.
Pediatric Patients 6 Years of Age and Older Who Also Weigh at Least 40 kg
The recommended dose is 100 mg.
2.3 Recommended Dosage for Active Psoriatic Arthritis
Administer TREMFYA by subcutaneous injection at Week 0, Week 4, and every 8 weeks thereafter.
TREMFYA may be administered alone or in combination with a conventional disease-modifying antirheumatic drug (e.g., methotrexate).
Adults
The recommended dose is 100 mg.
Pediatric Patients 6 Years of Age and Older Who Also Weigh at Least 40 kg
The recommended dose is 100 mg.
2.4 Recommended Dosage for Moderately to Severely Active Ulcerative Colitis and Crohn's Disease
Adults
Induction:
The recommended induction dosage of TREMFYA is:
- 200 mg administered by intravenous infusion over at least one hour at Week 0, Week 4, and Week 8 [see Dosage and Administration (2.6)] or
- 400 mg administered by subcutaneous injection (given as two consecutive injections of 200 mg each) at Week 0, Week 4, and Week 8.
Maintenance:
The recommended maintenance dosage of TREMFYA is:
- 100 mg administered by subcutaneous injection at Week 16, and every 8 weeks thereafter, or
- 200 mg administered by subcutaneous injection at Week 12, and every 4 weeks thereafter.
Use the lowest effective recommended dosage to maintain therapeutic response.
2.5 Preparation and Administration Instructions for Subcutaneous Injection
TREMFYA is available for subcutaneous use in the following presentations: prefilled pen (TREMFYA PEN), One-Press injector, and prefilled syringes [see Dosage Forms and Strengths (3) and How Supplied/Storage and Handling (16)].
- Each prefilled pen, One-Press injector, or prefilled syringe is for one time use in one patient only. Instruct patients to inject the full amount: 100 mg or 200 mg of TREMFYA (1 mL or 2 mL, respectively).
-
TREMFYA is intended for use under the guidance and supervision of a healthcare professional. After proper training in subcutaneous injection technique:
Adults- Adults may self-inject with the TREMFYA prefilled syringe, One-Press injector, and prefilled pen.
- Inject into the front of the thighs, the lower abdomen except for the 2 inches around the navel, or the back of the upper arms (healthcare professional or caregiver only).
- Pediatric self-administration is not recommended. Administration of TREMFYA to pediatric patients with the prefilled syringe, One-Press injector and prefilled pen should be performed by a healthcare provider or by a caregiver who has received training and demonstrated proper subcutaneous injection technique.
- Inject into the front of the thighs or the lower abdomen except for the 2 inches around the navel. For the prefilled syringe only, injection can also be given in the back of the upper arms.
- Before injection, remove TREMFYA from the refrigerator and allow to reach room temperature up to 25 °C (77 °F) (30 minutes) without removing the needle cap.
- Do not inject TREMFYA into areas where the skin is tender, bruised, red, hard, thick, scaly, or affected by psoriasis [see Instructions for Use].
- The TREMFYA Instructions for Use contains more detailed patient instructions on the preparation and administration of TREMFYA [see Instructions for Use].
- If a dose is missed, administer the dose as soon as possible. Thereafter, resume dosing at the regular scheduled time.
- Inspect TREMFYA visually for particulate matter and discoloration prior to administration. TREMFYA is a clear and colorless to light yellow solution that may contain small translucent particles. Do not use if the liquid contains large particles, is discolored or cloudy. TREMFYA does not contain preservatives; therefore, discard any unused product remaining in the prefilled pen, One-Press injector, or prefilled syringe.
2.6 Preparation and Administration Instructions for Intravenous Infusion (Moderately to Severely Active Ulcerative Colitis and Crohn's Disease)
Preparation Instructions:
- Withdraw and then discard 20 mL of the 0.9% Sodium Chloride Injection from the 250 mL infusion bag which is equal to the volume of TREMFYA to be added.
- Withdraw 20 mL of TREMFYA from the vial and add it to the 250 mL intravenous infusion bag of 0.9% Sodium Chloride Injection for a final concentration of 0.8 mg/mL. Gently mix the diluted solution. Discard the vial with any remaining solution.
- Visually inspect the diluted solution for particulate matter and discoloration before infusion. Infuse the diluted solution over a period of at least one hour.
- Use only an infusion set with an in-line, sterile, non-pyrogenic, low protein binding filter (pore size 0.2 micrometer).
- Do not infuse TREMFYA concomitantly in the same intravenous line with other medicinal products.
- Dispose any unused medicinal product in accordance with local requirements.
Administration Instructions:
- TREMFYA solution for intravenous infusion must be diluted, prepared, and infused by a healthcare professional using aseptic technique. TREMFYA does not contain preservatives. Each vial is for one time use in one patient only.
- Inspect TREMFYA visually for particulate matter and discoloration prior to administration. TREMFYA is a clear and colorless to light yellow solution that may contain small translucent particles. Do not use if the liquid contains large particles, is discolored, or is cloudy.
Storage of Diluted Solution:
- The diluted infusion solution may be kept at room temperature up to 25 °C (77 °F) for up to 10 hours. Storage time at room temperature begins once the diluted solution has been prepared. The infusion should be completed within 10 hours after the dilution in the infusion bag.
- Do not freeze.
- Discard any unused portion of the infusion solution.
3 DOSAGE FORMS AND STRENGTHS
TREMFYA is a clear and colorless to light yellow solution.
Subcutaneous Injection (3)
- Injection: 100 mg/mL in a single-dose One-Press patient-controlled injector.
- Injection: 100 mg/mL in a single-dose prefilled pen (TREMFYA PEN).
- Injection: 200 mg/2 mL in a single-dose prefilled pen (TREMFYA PEN).
- Injection: 100 mg/mL in a single-dose prefilled syringe.
- Injection: 200 mg/2 mL (100 mg/mL) in a single-dose prefilled syringe.
Intravenous Infusion (3)
- Injection: 200 mg/20 mL (10 mg/mL) solution in a single-dose vial.
Subcutaneous Injection
- Injection: 100 mg/mL in a single-dose One-Press patient-controlled injector.
- Injection: 100 mg/mL in a single-dose prefilled pen (TREMFYA PEN).
- Injection: 200 mg/2 mL in a single-dose prefilled pen (TREMFYA PEN).
- Injection: 100 mg/mL in a single-dose prefilled syringe.
- Injection: 200 mg/2 mL (100 mg/mL) in a single-dose prefilled syringe.
Intravenous Infusion
- Injection: 200 mg/20 mL (10 mg/mL) solution in a single-dose vial.
4 CONTRAINDICATIONS
TREMFYA is contraindicated in patients with a history of serious hypersensitivity reaction to guselkumab or to any of the excipients [see Warnings and Precautions (5.1)].
Serious hypersensitivity reactions to guselkumab or to any of the excipients. (4)
5 WARNINGS AND PRECAUTIONS
- Hypersensitivity Reactions: Serious hypersensitivity reactions, including anaphylaxis, may occur. (5.1)
- Infections: TREMFYA may increase the risk of infections. Do not initiate treatment with TREMFYA in patients with clinically important active infection until the infection resolves or is adequately treated. If such an infection develops, discontinue TREMFYA until the infection resolves. (5.2)
- Tuberculosis (TB): Evaluate for TB prior to initiating treatment with TREMFYA. Monitor patients for signs and symptoms of active TB during and after treatment with TREMFYA. (5.3)
- Hepatotoxicity: Drug-induced liver injury has been reported. For the treatment of ulcerative colitis or Crohn’s disease, evaluate liver enzymes and bilirubin levels at baseline, for at least 16 weeks of treatment, and periodically thereafter according to routine patient management. For the treatment of plaque psoriasis or psoriatic arthritis, if clinically indicated, evaluate liver enzymes and bilirubin at baseline, and periodically thereafter according to routine patient management. Interrupt treatment if drug-induced liver injury is suspected, until this diagnosis is excluded. (5.4)
- Immunizations: Avoid use of live vaccines. (5.5)
5.1 Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylaxis, have been reported with post market use of TREMFYA. Some cases required hospitalization. If a serious hypersensitivity reaction occurs, discontinue TREMFYA and initiate appropriate therapy.
5.2 Infections
TREMFYA may increase the risk of infection [see Adverse Reactions (6.1)].
In placebo-controlled clinical trials of up to 48 weeks in subjects with ulcerative colitis and Crohn’s disease, serious infections occurred in ≤ 2% of subjects who received TREMFYA. In the 16-week placebo-controlled trials in subjects with plaque psoriasis, the rate of serious infections for the TREMFYA group and the placebo group was ≤ 0.2%. A similar rate of serious infections was seen in placebo-controlled trials in subjects with psoriatic arthritis. The overall rates of infections were similar between subjects in the TREMFYA groups and subjects in the placebo groups in clinical trials for all indicated populations [see Adverse Reactions (6.1)].
Treatment with TREMFYA should not be initiated in patients with any clinically important active infection until the infection resolves or is adequately treated.
In patients with a chronic infection or a history of recurrent infection, consider the risks and benefits prior to prescribing TREMFYA. Instruct patients to seek medical help if signs or symptoms of clinically important chronic or acute infection occur. If a patient develops a clinically important or serious infection or is not responding to standard therapy, monitor the patient closely and discontinue TREMFYA until the infection resolves.
5.3 Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating TREMFYA treatment. Do not administer TREMFYA to patients with active TB infection. Initiate treatment of latent TB prior to administering TREMFYA. Consider anti-TB therapy prior to initiating TREMFYA in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Monitor all patients for signs and symptoms of active TB during and after TREMFYA treatment.
In clinical trials, 105 subjects with plaque psoriasis, 71 subjects with psoriatic arthritis, 43 subjects with ulcerative colitis, and 36 subjects with Crohn's disease with latent TB who were concurrently treated with TREMFYA, and appropriate TB prophylaxis did not develop active TB. In clinical trials of TREMFYA in subjects with Crohn's disease, active TB was reported in 2 subjects during treatment with TREMFYA [see Adverse Reactions (6.1)].
5.4 Hepatotoxicity
A serious adverse reaction of drug-induced liver injury was reported in a clinical trial subject with Crohn's disease following three doses of a higher than the recommended induction regimen. This subject had peak alanine aminotransferase (ALT) of 18x the upper limit of normal (ULN), aspartate aminotransferase (AST) of 11x ULN, and total bilirubin of 2.4x ULN. TREMFYA was subsequently discontinued, and the liver test abnormalities resolved following administration of corticosteroids [see Adverse Reactions (6.1)].
In patients with ulcerative colitis or Crohn's disease, evaluate liver enzymes and bilirubin at baseline, for at least 16 weeks of treatment, and periodically thereafter according to routine patient management.
In patients with plaque psoriasis or psoriatic arthritis, if clinically indicated, evaluate liver enzymes and bilirubin at baseline, and periodically thereafter according to routine patient management.
Consider other treatment options in patients with evidence of acute liver disease or cirrhosis. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury. Interrupt treatment if drug-induced liver injury is suspected, until this diagnosis is excluded. Instruct patients to seek immediate medical attention if they experience symptoms suggestive of hepatic dysfunction.
5.5 Immunizations
Avoid use of live vaccines in patients treated with TREMFYA. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Prior to initiating therapy with TREMFYA, complete all age-appropriate vaccinations according to current immunization guidelines. No data are available on the response to live or inactive vaccines.
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of labeling:
- Hypersensitivity Reactions [see Contraindications (4) and Warnings and Precautions (5.1)]
- Infections [see Warnings and Precautions (5.2)]
- Tuberculosis [see Warnings and Precautions (5.3)]
- Hepatotoxicity [see Warnings and Precautions (5.4)]
Most common adverse reactions associated with TREMFYA are:
- Plaque Psoriasis and Psoriatic Arthritis (≥1%): upper respiratory infections, headache, injection site reactions, arthralgia, bronchitis, diarrhea, gastroenteritis, tinea infections, and herpes simplex infections. (6.1)
- Ulcerative Colitis (≥3%): injection site reactions, arthralgia, upper respiratory tract infections, headache, gastroenteritis, fatigue, pyrexia, and rash. (6.1)
- Crohn's Disease (≥3%): respiratory tract infections, abdominal pain, injection site reactions, headache, fatigue, arthralgia, diarrhea, and gastroenteritis. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Biotech, Inc. at 1-800-526-7736 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Adults with Plaque Psoriasis
In clinical trials, a total of 1823 adult subjects with moderate-to-severe plaque psoriasis received TREMFYA. Of these, 1393 subjects were exposed to TREMFYA for at least 6 months and 728 subjects were exposed for at least 1 year.
Data from two placebo- and active-controlled trials (PsO1 and PsO2) in 1441 subjects (mean age 44 years; 70% males; 82% white) were pooled to evaluate the safety of TREMFYA (100 mg administered subcutaneously at Weeks 0 and 4, followed by every 8 weeks).
Weeks 0 to 16:
In the 16-week placebo-controlled period of the pooled clinical trials (PsO1 and PsO2), adverse events occurred in 49% of subjects in the TREMFYA group compared to 47% of subjects in the placebo group and 49% of subjects in the U.S. licensed adalimumab group. Serious adverse events occurred in 1.9% of subjects in the TREMFYA group (6.3 events per 100 patient-years of follow-up) compared to 1.4% of subjects in the placebo group (4.7 events per 100 patient-years of follow-up), and in 2.6% of subjects in U.S. licensed adalimumab group (9.9 events per 100 patient-years of follow-up).
Table 1 summarizes the adverse reactions that occurred at a rate of at least 1% and at a higher rate in the TREMFYA group than in the placebo group during the 16-week placebo-controlled period.
Adverse reactions that occurred in < 1% but > 0.1% of subjects in the TREMFYA group and at a higher rate than in the placebo group through Week 16 in trials PsO1 and PsO2 were migraine, candida infections, and urticaria.
Specific Adverse Reactions
- Infections
Infections occurred in 23% of subjects in the TREMFYA group compared to 21% of subjects in the placebo group through 16 weeks of treatment.
The most common (≥ 1%) infections that occurred more frequently in the TREMFYA group than in the placebo group were upper respiratory infections, gastroenteritis, tinea infections, and herpes simplex infections; all cases were mild to moderate in severity and did not lead to discontinuation of TREMFYA. The rate of serious infections for the TREMFYA group and the placebo group was ≤ 0.2%.
-
Elevated Liver Enzymes
Elevated liver enzymes were reported more frequently in the TREMFYA group (2.6%) than in the placebo group (1.9%). Of the 21 subjects who were reported to have elevated liver enzymes in the TREMFYA group, all events except one were mild to moderate in severity and none of the events led to discontinuation of TREMFYA.
-
Safety through Week 48
Through Week 48, no new adverse reactions were identified with TREMFYA use and the frequency of the adverse reactions was similar to the safety profile observed during the first 16 weeks of treatment.
Adverse Reactions in Pediatric Subjects with Plaque Psoriasis
The safety of TREMFYA was studied in one placebo- and active-controlled clinical trial (PsO5) that included 120 pediatric subjects 6 years of age and older with moderate-to-severe plaque psoriasis [see Clinical Studies (14.1)]. The safety profile observed in pediatric subjects 6 years of age and older treated with TREMFYA up to 52 weeks was consistent with the safety profile observed in adult subjects with moderate-to-severe plaque psoriasis.
Adverse Reactions in Adults with Psoriatic Arthritis
TREMFYA was studied in two placebo-controlled trials in adult subjects with psoriatic arthritis (748 subjects on TREMFYA and 372 subjects on placebo). Of the 748 subjects who received TREMFYA, 375 subjects received TREMFYA 100 mg at Week 0, Week 4, and every 8 weeks thereafter and 373 subjects received TREMFYA 100 mg every 4 weeks. The overall safety profile observed in adult subjects with psoriatic arthritis treated with TREMFYA is generally consistent with the safety profile in adult subjects with plaque psoriasis with the addition of bronchitis and neutrophil count decreased. In the 24-week placebo-controlled period, combined across the two studies, bronchitis occurred in 1.6% of subjects in the TREMFYA q8w group and 2.9% of subjects in the TREMFYA q4w group compared to 1.1% of subjects in the placebo group. Neutrophil count decreased occurred in 0.3% of subjects in the TREMFYA q8w and 1.6% of subjects in the TREMFYA q4w group compared to 0% of subjects in the placebo group. The majority of events of neutrophil count decreased were mild, transient, not associated with infection and did not lead to discontinuation. A similar risk of infection and serious infections was seen in placebo-controlled trials in subjects with psoriatic arthritis as in subjects with plaque psoriasis.
Adverse Reactions in Adults with Ulcerative Colitis
TREMFYA was studied up to 12 weeks in adult subjects with moderately to severely active ulcerative colitis in a randomized, double-blind, placebo-controlled induction trial (UC1) and a randomized, double-blind, placebo-controlled, induction dose-finding trial (UC3; NCT04033445). Long-term safety up to 44 weeks was evaluated in subjects who responded to induction therapy in trials UC1 or UC3 in a randomized, double-blind, placebo-controlled maintenance trial (UC2). In a randomized, double-blind, placebo-controlled trial (UC4), subjects received subcutaneous TREMFYA at Weeks 0, 4, and 8 followed by one of two recommended subcutaneous TREMFYA dosing regimens for a total duration of up to 24 weeks [see Clinical Studies (14.3)].
Trials UC1, UC2 and UC3
In the induction trials (UC1 and UC3), 522 subjects received at least one dose of the TREMFYA intravenous induction regimen (i.e., 200 mg at Week 0, Week 4, and Week 8). Clinical response was defined as a decrease in modified Mayo score (mMS) of ≥30% and ≥2 points from baseline with either a ≥1 decrease from baseline in rectal bleeding subscore (RBS) or RBS of 0 or 1. In the maintenance trial (UC2), subjects who achieved clinical response after 12 weeks of TREMFYA intravenous induction treatment were randomized and received either TREMFYA 100 mg every 8 weeks (with the first dose given at Week 4 of trial UC2) or TREMFYA 200 mg every 4 weeks (with the first dose given at Week 0 of trial UC2), by subcutaneous (SC) injection for up to an additional 44 weeks.
Respiratory tract infections occurred in ≥2% of subjects treated with TREMFYA and at a higher rate than placebo (8.8% TREMFYA-treated subjects vs. 7.3% placebo-treated subjects) through Week 12 in the induction trials (UC1 and UC3). Respiratory tract infections included COVID-19, influenza, nasopharyngitis, respiratory tract infection, upper respiratory tract infection, and viral respiratory tract infection.
Adverse reactions that occurred in ≥3% of subjects treated with TREMFYA and at a higher rate than placebo through Week 44 in the maintenance trial (UC2) are shown in Table 2.
Trial UC4
In trial UC4, 418 subjects were enrolled, of whom 279 subjects were randomized to receive TREMFYA. Randomization was 1:1:1 to subcutaneous TREMFYA 400 mg at Weeks 0, 4, and 8 followed by subcutaneous TREMFYA 100 mg every 8 weeks (with the first dose given at Week 16) for up to an additional 8 weeks (from Week 16 through Week 24); subcutaneous TREMFYA 400 mg at Weeks 0, 4, and 8 followed by subcutaneous TREMFYA 200 mg every 4 weeks (with the first dose given at Week 12 for up to an additional 12 weeks (from Week 12 through Week 24); or placebo.
Adverse reactions reported in trial UC4 through Week 24 in ≥3% of subjects treated with either subcutaneous TREMFYA dosing regimen and at a higher rate than placebo are shown in Table 3.
Specific Adverse Reactions
Infections
In the 44-week trial (UC2), serious infections occurred in 0.8% of subjects treated with TREMFYA versus 0% of subjects in the placebo group. In the 24-week trial (UC4), serious infections occurred in 1.8% of subjects treated with TREMFYA versus 0.7% of subjects in the placebo group.
Adverse Reactions in Adults with Crohn's Disease
TREMFYA was studied in four clinical trials in adult subjects with moderately to severely active Crohn's disease [see Clinical Studies (14.4)]. In two randomized, double-blind, placebo-controlled trials (CD1, CD2) and one randomized, double-blind, dose-ranging trial (CD4, NCT03466411); subjects received intravenous TREMFYA at Weeks 0, 4, and 8 followed by one of two recommended subcutaneous TREMFYA dosing regimens for a total duration of up to 48 weeks. In a randomized, double-blind, placebo-controlled trial (CD3); subjects received subcutaneous TREMFYA at Weeks 0, 4, and 8 followed by one of two recommended subcutaneous TREMFYA dosing regimens for a total duration of up to 48 weeks.
Trials CD1, CD2, and CD4
In trials CD1, CD2, and CD4; 1349 subjects were enrolled, of whom 649 were randomized to intravenous TREMFYA 200 mg at Weeks 0, 4, and 8 followed by either subcutaneous TREMFYA 100 mg every 8 weeks (with the first dose given at Week 16) for up to an additional 32 weeks (from Week 16 through Week 48) or subcutaneous TREMFYA 200 mg every 4 weeks (with the first dose given at Week 12) for up to an additional 36 weeks (from Week 12 through Week 48); and 211 subjects were randomized to receive placebo.
Through Week 12 in trials CD1, CD2, and CD4; headache (including headache, migraine, and sinus headache) was reported in ≥3% of subjects treated with intravenous TREMFYA and at a greater rate than placebo (3.4% TREMFYA-treated subjects vs. 1.9% placebo-treated subjects). In TREMFYA-treated subjects in trial CD2, one case of active extrapulmonary TB was reported between Weeks 12–48 and one case of active pulmonary TB was reported after Week 48. Both subjects had negative baseline screenings and resided in TB-endemic regions [see Warnings and Precautions (5.3)]. In general, the safety profile in subjects treated with subcutaneous TREMFYA from Week 12 up to Week 48 in these trials was generally similar to that reported with intravenous TREMFYA through Week 12.
Trial CD3
In trial CD3, 347 subjects were enrolled, of whom 230 subjects were randomized to receive TREMFYA. Randomization was 1:1:1 to subcutaneous TREMFYA 400 mg at Weeks 0, 4, and 8 followed by subcutaneous TREMFYA 100 mg every 8 weeks (with the first dose given at Week 16) for up to an additional 32 weeks (from Week 16 through Week 48); subcutaneous TREMFYA 400 mg at Weeks 0, 4, and 8 followed by subcutaneous TREMFYA 200 mg every 4 weeks (with the first dose given at Week 12) for up to an additional 36 weeks (from Week 12 through Week 48); or placebo.
Adverse reactions reported in trial CD3 through Week 48 in ≥3% of subjects treated with either subcutaneous TREMFYA dosing regimen and at a higher rate than placebo are shown in Table 4.
Specific Adverse Reactions
Infections
In the 48-week clinical trial in subjects with Crohn’s disease (CD3), serious infections occurred in 1.5% of subjects treated with TREMFYA versus 0% of subjects in the placebo group.
Elevated Liver Enzymes
Through Week 12 in CD1, CD2, and CD4; ALT ≥5× ULN was reported in 2/645 (0.3%) subjects treated with intravenous TREMFYA 200 mg at Weeks 0, 4, and 8 and 0/211 subjects treated with placebo. These elevations occurred without concomitant elevations in total bilirubin.
Through Week 48 in CD3, ALT ≥5× ULN was reported in 0/115 subjects treated with subcutaneous TREMFYA 400 mg at Weeks 0, 4, and 8 followed by subcutaneous TREMFYA 100 mg every 8 weeks; 2/115 (1.7%) subjects treated with subcutaneous TREMFYA 400 mg at Weeks 0, 4, and 8 followed by subcutaneous TREMFYA 200 mg every 4 weeks; and 0/117 subjects treated with placebo. These elevations occurred without concomitant elevations in total bilirubin.
6.2 Postmarketing Experience
The following adverse reactions have been reported during post-approval of TREMFYA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to TREMFYA exposure.
Immune system disorders: Hypersensitivity, including anaphylaxis [see Warnings and Precautions (5.1)]
Skin and subcutaneous tissue disorders: Rash [see Warnings and Precautions (5.1)]
7 DRUG INTERACTIONS
7.1 CYP450 Substrates
The formation of CYP450 enzymes can be altered by increased levels of certain cytokines (e.g., IL-1, IL-6, IL-10, TNFα, interferon) during chronic inflammation.
Results from an exploratory drug-drug interaction trial in subjects with moderate-to-severe plaque psoriasis suggested a low potential for clinically relevant drug interactions for drugs metabolized by CYP3A4, CYP2C9, CYP2C19 and CYP1A2 but the interaction potential cannot be ruled out for drugs metabolized by CYP2D6. However, the results were highly variable because of the limited number of subjects in the trial.
Upon initiation of TREMFYA in patients who are receiving concomitant CYP450 substrates, particularly those with a narrow therapeutic index, consider monitoring for therapeutic effect or drug concentration and consider dosage adjustment as needed [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy registry that monitors pregnancy outcomes in women exposed to TREMFYA during pregnancy. Patients should be encouraged to enroll in the registry by visiting www.mothertobaby.org/ongoing-study/tremfya-guselkumab, by calling 1-877-311-8972, or emailing MotherToBaby@health.ucsd.edu.
Risk Summary
Available data from literature, post-marketing reports, and ongoing pregnancy registry with TREMFYA use in pregnant women are insufficient to establish a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. Monoclonal antibodies are actively transported across the placenta (see Clinical Considerations).
In a combined embryofetal development and pre- and post-natal development study, no adverse developmental effects were observed in infants born to pregnant monkeys after subcutaneous administration of guselkumab during organogenesis through parturition at doses up to 18 times the exposure (AUC) in humans administered 200 mg intravenously and 16 times the exposure (AUC) to the 400 mg dose given subcutaneously. Neonatal deaths in monkeys were observed at 4 to 18 times the exposure (AUC) in humans administered 200 mg intravenously and 4 to 16 times the exposure (AUC) to the 400 mg dose given subcutaneously (see Data). The clinical significance of these nonclinical findings is unknown.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated Maternal and Embryo/Fetal Risk
Published data suggest that the risk of adverse pregnancy outcomes in women with inflammatory bowel disease (IBD) is associated with increased disease activity. Adverse pregnancy outcomes include preterm delivery (before 37 weeks gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Fetal/Neonatal Adverse Reactions
Transport of endogenous IgG antibodies across the placenta increases as pregnancy progresses and peaks during the third trimester. Therefore, TREMFYA may be present in infants exposed in utero. The potential clinical impact of guselkumab exposure in infants exposed in utero should be considered.
Data
Animal Data
In a combined embryofetal development and pre- and post-natal development study, pregnant cynomolgus monkeys were administered weekly subcutaneous doses of guselkumab from the beginning of organogenesis to parturition at a dose (50 mg/kg) resulting in exposures (AUC) 18 times the exposure in humans administered 200 mg intravenously and 16 times the human exposure at 400 mg given subcutaneously. Neonatal deaths occurred in the offspring of one control monkey, three monkeys administered guselkumab at 10 mg/kg/week (4 times the exposure (AUC) in humans administered 200 mg intravenously or 400 mg given subcutaneously) and three monkeys administered guselkumab at 50 mg/kg/week (18 times the exposure (AUC) in humans administered 200 mg intravenously and 16 times the exposure (AUC) following a 400 mg subcutaneous dose). The clinical significance of these findings is unknown. No guselkumab-related effects on functional or immunological development were observed in the infants from birth through 6 months of age.
8.2 Lactation
Risk Summary
There are no data on the presence of guselkumab in human milk, the effects on the breastfed infant, or the effects on milk production. Guselkumab was not detected in the milk of lactating cynomolgus monkeys. Endogenous maternal IgG and monoclonal antibodies are transferred into human milk. The effects of local gastrointestinal exposure and the extent of systemic exposure in the breastfed infant to guselkumab are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TREMFYA and any potential adverse effects on the breastfed infant from TREMFYA or from the underlying maternal condition.
8.4 Pediatric Use
Plaque Psoriasis
The safety and effectiveness of TREMFYA for the treatment of moderate-to-severe plaque psoriasis have been established in pediatric patients 6 years of age and older who are candidates for systemic therapy or phototherapy [see Adverse Reactions (6.1) and Clinical Studies (14.1)].
The safety and effectiveness of TREMFYA have not been established in pediatric patients with plaque psoriasis who are younger than 6 years of age.
Psoriatic Arthritis
The safety and effectiveness of TREMFYA have been established for treatment of active psoriatic arthritis in pediatric patients 6 years of age and older.
Use of TREMFYA in this age group is supported by evidence from adequate and well controlled trials of TREMFYA in adults with plaque psoriasis and psoriatic arthritis, pharmacokinetic data from adult subjects with plaque psoriasis and psoriatic arthritis and pediatric subjects with plaque psoriasis, and safety data from a clinical trial in 120 subjects 6 to 17 years of age with plaque psoriasis. The observed pre-dose (trough) concentrations are generally comparable between adult subjects with plaque psoriasis, adult subjects with psoriatic arthritis and pediatric subjects with plaque psoriasis, and the systemic exposure is expected to be comparable between adult and pediatric patients with psoriatic arthritis [see Adverse Reactions (6.1) , Clinical Pharmacology (12.3) , and Clinical Studies (14.1, 14.2)].
The safety and effectiveness of TREMFYA have not been established in pediatric patients with psoriatic arthritis who are younger than 6 years of age.
Ulcerative Colitis
The safety and effectiveness of TREMFYA have not been established in pediatric patients with ulcerative colitis.
Crohn's Disease
The safety and effectiveness of TREMFYA have not been established in pediatric patients with Crohn’s disease.
8.5 Geriatric Use
Of the 5723 subjects with plaque psoriasis, psoriatic arthritis, ulcerative colitis, or Crohn's disease exposed to TREMFYA, a total of 313 subjects were 65 years or older, and 32 subjects were 75 years or older. Clinical studies of TREMFYA, within each indication, did not include sufficient numbers of subjects 65 years of age and older to determine whether they respond differently from younger adult subjects.
No clinically meaningful differences in the pharmacokinetics of guselkumab were observed based on age [see Clinical Pharmacology (12.3)].
11 DESCRIPTION
Guselkumab, an interleukin-23 antagonist, is a human immunoglobulin G1 lambda (IgG1λ) monoclonal antibody. Guselkumab is produced in a mammalian cell line using recombinant DNA technology and has an approximate molecular weight of approximately 147 kDa.
TREMFYA® (guselkumab) injection is a sterile, preservative free, clear, and colorless to light yellow solution.
TREMFYA for Subcutaneous Injection
Available as a 100 mg/mL solution in a 1 mL or 2 mL glass prefilled syringe, in a 1 mL or 2 mL prefilled pen (TREMFYA PEN), or in a 1 mL One-Press patient-controlled injector for subcutaneous use.
- Each TREMFYA 1 mL prefilled syringe, prefilled pen (TREMFYA PEN), or One-Press patient-controlled injector delivers 100 mg guselkumab, L-histidine (0.6 mg), L-histidine monohydrochloride monohydrate (1.5 mg), polysorbate 80 (0.5 mg), sucrose (79 mg) and water for injection at pH 5.8.
- Each TREMFYA 2 mL prefilled syringe or prefilled pen (TREMFYA PEN) delivers 200 mg guselkumab, L-histidine (1.2 mg), L-histidine monohydrochloride monohydrate (3 mg), polysorbate 80 (1 mg), sucrose (158 mg) and water for injection at pH 5.8.
TREMFYA for Intravenous Infusion
Available as 10 mg/mL solution in a 20 mL single-dose vial for intravenous use.
Each TREMFYA 20 mL vial delivers 200 mg guselkumab, EDTA disodium dihydrate (0.4 mg), L-histidine (11.3 mg), L-histidine monohydrochloride monohydrate (26.6 mg), L-methionine (8 mg), polysorbate 80 (10 mg), sucrose (1700 mg) and water for injection at pH 5.8.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Guselkumab is a human monoclonal IgG1λ antibody that selectively binds to the p19 subunit of interleukin 23 (IL-23) and inhibits its interaction with the IL-23 receptor. IL-23 is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. Guselkumab inhibits the release of proinflammatory cytokines and chemokines.
12.2 Pharmacodynamics
In evaluated adult subjects with moderate-to-severe plaque psoriasis, guselkumab reduced serum levels of IL-17A, IL-17F and IL-22 relative to pre-treatment levels based on exploratory analyses of the pharmacodynamic markers.
In evaluated adult subjects with active psoriatic arthritis, serum levels of acute phase proteins C-reactive protein, serum amyloid A and IL-6, and Th17 effector cytokines IL-17A, IL-17F and IL-22 were elevated at baseline. Serum levels of these proteins measured at Week 4 and Week 24 were decreased compared to baseline following guselkumab treatment at Week 0, Week 4, and every 8 weeks thereafter.
The relationship between these pharmacodynamic markers and the mechanism(s) by which guselkumab exerts its clinical effects is unknown.
12.3 Pharmacokinetics
Guselkumab exhibited linear pharmacokinetics in healthy adult subjects and adult subjects with plaque psoriasis following subcutaneous injections. In subjects with plaque psoriasis, following subcutaneous administration of 100 mg of TREMFYA at Weeks 0 and 4, and every 8 weeks thereafter, mean steady-state trough serum guselkumab concentration was approximately 1.2 mcg/mL.
The pharmacokinetics of guselkumab in adult subjects with psoriatic arthritis was similar to that in adult subjects with plaque psoriasis. Following subcutaneous administration of 100 mg of TREMFYA at Weeks 0, 4, and every 8 weeks thereafter, mean steady-state trough serum guselkumab concentration was approximately 1.2 mcg/mL.
Following subcutaneous maintenance dosing of 100 mg TREMFYA every 8 weeks or 200 mg TREMFYA every 4 weeks in adult subjects with ulcerative colitis, mean steady-state trough serum guselkumab concentrations were approximately 1.4 mcg/mL and 10.7 mcg/mL, respectively.
Following subcutaneous maintenance dosing of 100 mg TREMFYA every 8 weeks, or 200 mg TREMFYA every 4 weeks, in adult subjects with Crohn's disease mean steady-state trough serum guselkumab concentrations were approximately 1.2 mcg/mL and 10.1 mcg/mL, respectively.
Absorption
Following a single 100 mg subcutaneous injection in healthy adult subjects, guselkumab reached a mean (± SD) maximum serum concentration of 8.09 ± 3.68 mcg/mL by approximately 5.5 days post dose. The absolute bioavailability of guselkumab following a single 100 mg subcutaneous injection was estimated to be approximately 49% in healthy adult subjects.
Following the recommended intravenous induction dose regimen of TREMFYA 200 mg at Weeks 0, 4, and 8, mean (± SD) peak serum guselkumab concentration at Week 8 was 68.3 ± 17.3 mcg/mL in adult subjects with ulcerative colitis.
Following the recommended subcutaneous induction dose regimen of TREMFYA 400 mg at Weeks 0, 4, and 8, mean (± SD) peak serum guselkumab concentration was estimated to be 28.8 ± 8.8 mcg/mL in adult subjects with ulcerative colitis. The total systemic exposure (AUC) after the recommended induction dose regimen was similar following subcutaneous and intravenous induction.
Following the recommended intravenous induction dose regimen of TREMFYA 200 mg at Weeks 0, 4, and 8, mean (± SD) peak serum guselkumab concentration at Week 8 was 70.5 ± 24.5 mcg/mL in adult subjects with Crohn's disease.
Following the recommended subcutaneous induction dose regimen of TREMFYA 400 mg at Weeks 0, 4, and 8, mean (± SD) peak serum guselkumab concentration was estimated to be 27.7 ± 9.1 mcg/mL in adult subjects with Crohn's disease. The total systemic exposure (AUC) after the recommended induction dose regimen was similar following subcutaneous and intravenous induction.
Distribution
In adult subjects with plaque psoriasis, apparent volume of distribution was 13.5 L. In adult subjects with ulcerative colitis, apparent volume of distribution at steady-state was 10.1 L. In adult subjects with Crohn's disease, apparent volume of distribution at steady-state was 11.4 L.
Elimination
Apparent clearance in adult subjects with plaque psoriasis was 0.516 L/day. Mean half-life of guselkumab was approximately 15 to 18 days in subjects with plaque psoriasis across trials.
The apparent clearance in adult subjects with ulcerative colitis was 0.531 L/day. Mean half-life of guselkumab was approximately 17 days in subjects with ulcerative colitis.
The apparent clearance in adult subjects with Crohn's disease was 0.568 L/day. Mean half-life of guselkumab was approximately 17 days in subjects with Crohn's disease.
Metabolism
The exact pathway through which guselkumab is metabolized has not been characterized. As a human IgG monoclonal antibody, guselkumab is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG.
Specific Populations
Geriatric Subjects
No apparent differences in clearance were observed in subjects ≥ 65 years of age compared to subjects < 65 years of age, suggesting no dose adjustment is needed for elderly subjects. Clearance and volume of distribution of guselkumab increases as body weight increases, however, observed clinical trial data indicate that dose adjustment for body weight is not warranted. No specific trials have been conducted to determine the effect of renal or hepatic impairment on the pharmacokinetics of guselkumab.
Pediatric Subjects
In trial PsO5, steady-state serum concentrations of guselkumab were achieved by Week 20 in pediatric subjects 6 years of age and older with moderate-to-severe plaque psoriasis.
Overall, the observed guselkumab trough concentrations in pediatric subjects 6 years of age and older and also weighing at least 40 kg with plaque psoriasis were within range of those observed for adult subjects with plaque psoriasis and adult subjects with psoriatic arthritis after administration of TREMFYA.
Drug Interactions
Population pharmacokinetic analyses indicated that concomitant use of NSAIDs, oral corticosteroids and conventional DMARDs such as methotrexate (MTX), azathioprine (AZA), and 6-mercaptopurine (6-MP), did not affect the clearance of guselkumab.
Cytochrome P450 Substrates
The effects of guselkumab on the pharmacokinetics of midazolam (metabolized by CYP3A4), warfarin (metabolized by CYP2C9), omeprazole (metabolized by CYP2C19), dextromethorphan (metabolized by CYP2D6), and caffeine (metabolized by CYP1A2) were evaluated in an exploratory study with 6 to 12 evaluable subjects with moderate-to-severe plaque psoriasis. Changes in AUCinf of midazolam, S-warfarin, omeprazole, and caffeine after a single dose of guselkumab were not clinically relevant. For dextromethorphan, changes in AUCinf after guselkumab were not clinically relevant in 9 out of 10 subjects; however, a 2.9-fold change in AUCinf was observed in one individual [see Drug Interactions (7.1)].
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of guselkumab or of other guselkumab products.
Plaque Psoriasis
Adult Subjects
Up to Week 52, approximately 6% of adult subjects treated with TREMFYA developed antidrug antibodies. Of the subjects who developed antidrug antibodies, approximately 7% had antibodies that were classified as neutralizing antibodies. Among the 46 subjects who developed antibodies to guselkumab and had evaluable data, 21 subjects exhibited lower trough levels of guselkumab, including one subjects who experienced loss of efficacy after developing high antibody titers. Up to Week 156, approximately 9% of subjects treated with TREMFYA developed antidrug antibodies and of these subjects approximately 6% were classified as neutralizing antibodies. However, antibodies to guselkumab were generally not associated with changes in clinical response or development of injection-site reactions.
Pediatric Subjects
Up to Week 44, 18% (n=21/114) of pediatric subjects 6 years of age and older treated with TREMFYA developed anti-drug antibodies, none of the anti-drug antibodies were classified as neutralizing. The development of antibodies to guselkumab did not impact the pharmacokinetics of guselkumab and was not associated with changes in clinical response or development of injection-site reactions. However, the small number of subjects who were positive for antibodies to guselkumab limits definitive conclusions.
Psoriatic Arthritis
Up to Week 24, 2% (n=15) of adult subjects treated with TREMFYA developed antidrug antibodies. Of these subjects, 1 had antibodies that were classified as neutralizing antibodies. Overall, the small number of subjects who were positive for antibodies to guselkumab limits definitive conclusion of the effect of immunogenicity on the pharmacokinetics, efficacy, and safety of guselkumab.
Ulcerative Colitis
Up to Week 56 in trials UC1, UC2 and UC3, 11% (48/435) of adult subjects treated with TREMFYA at the recommended dosages developed antidrug antibodies. Of these subjects who tested positive for anti-guselkumab antibodies and were evaluable for neutralizing antibodies, 16% (6/38) had antibodies that were classified as neutralizing antibodies. Two subjects with the highest antibody titers exhibited low trough levels of guselkumab. Up to Week 24 in UC4, 9% (24/279) of subjects treated with TREMFYA developed antidrug antibodies. Of the subjects who tested positive for anti-guselkumab antibodies and were evaluable for neutralizing antibodies 13% (2/16) had antibodies that were classified as neutralizing antibodies. There was no identified clinically significant effect of antidrug antibodies on injection site reactions, or effectiveness of guselkumab, over the treatment duration of 56 weeks (trials UC1, UC2 and UC3) or 24 weeks (trial UC4).
Crohn's Disease
Up to Week 48 in trials CD1, CD2, and CD4; 5% (30/634) of adult subjects treated with a recommended dosage of TREMFYA developed antidrug antibodies. Of the subjects who tested positive for anti-guselkumab antibodies and were evaluable for neutralizing antibodies, 5% (1/22) had antibodies that were classified as neutralizing antibodies. Up to Week 48 in CD3, 9% (24/273) of subjects treated with TREMFYA developed antidrug antibodies. Of the subjects who tested positive for anti-guselkumab antibodies and were evaluable for neutralizing antibodies, none had antibodies that were classified as neutralizing antibodies. There were no identified clinically significant effects of antidrug antibodies on pharmacokinetics, injection site reactions, or effectiveness of guselkumab, over the treatment duration of 48 weeks.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Animal studies have not been conducted to evaluate the carcinogenic or mutagenic potential of TREMFYA.
No effects on fertility parameters were observed after male guinea pigs were subcutaneously administered guselkumab at a dose of 25 mg/kg twice weekly (6 times the exposure (AUC) in humans administered 200 mg intravenously and 5 times the exposure (AUC) at the 400 mg subcutaneous dose).
No effects on fertility parameters were observed after female guinea pigs were subcutaneously administered guselkumab at doses up to 100 mg/kg twice weekly (12 times the exposure (AUC) in humans administered 200 mg intravenously and 10 times the exposure (AUC) at the 400 mg subcutaneous dose).
14 CLINICAL STUDIES
14.1 Clinical Studies in Plaque Psoriasis
Adults with Moderate-to-Severe Plaque Psoriasis
Four multicenter, randomized, double-blind trials (PsO1 [NCT02207231], PsO2 [NCT02207244], PsO3 [NCT02203032], and PsO4 [NCT02905331]) enrolled subjects 18 years of age and older with moderate-to-severe plaque psoriasis who were eligible for systemic therapy or phototherapy. Subjects had an Investigator's Global Assessment (IGA) score of ≥3 ("moderate") on a 5-point scale of overall disease severity, a Psoriasis Area and Severity Index (PASI) score ≥12, and a minimum affected body surface area (BSA) of 10%. Subjects with guttate, erythrodermic, or pustular psoriasis were excluded.
Trials PsO1 and PsO2
In trials PsO1 and PsO2, 1443 subjects were randomized to either TREMFYA (100 mg at Weeks 0 and 4 and every 8 weeks thereafter) administered with a prefilled syringe, placebo or U.S. licensed adalimumab (80 mg at Week 0 and 40 mg at Week 1, followed by 40 mg every other week thereafter).
Both trials assessed the responses at Week 16 compared to placebo for the two co-primary endpoints:
- the proportion of subjects who achieved an IGA score of 0 ("cleared") or 1 ("minimal");
- the proportion of subjects who achieved at least a 90% reduction from baseline in the PASI composite score (PASI 90).
Comparisons between TREMFYA and U.S. licensed adalimumab were secondary endpoints at the following time points:
- at Week 16 (trials PsO1 and PsO2), the proportions of subjects who achieved an IGA score of 0 or 1, a PASI 90, and a PASI 75 response;
- at Week 24 (trials PsO1 and PsO2), and at Week 48 (trial PsO1), the proportions of subjects achieving an IGA score of 0, an IGA score of 0 or 1, and a PASI 90 response.
Other evaluated outcomes included improvement in psoriasis symptoms assessed on the Psoriasis Symptoms and Signs Diary (PSSD) and improvements in psoriasis of the scalp at Week 16.
In both trials, subjects were predominantly men and White, with a mean age of 44 years and a mean weight of 90 kg. At baseline, subjects had a median affected BSA of approximately 21%, a median PASI score of 19, and 18% had a history of psoriatic arthritis. Approximately 24% of subjects had an IGA score of 4 ("severe"). In both trials, 23% had received prior biologic systemic therapy.
Clinical Response
Table 5 presents the efficacy results at Week 16 in trials PsO1 and PsO2.
Table 6 presents the results of an analysis of all the North America sites (i.e., U.S. and Canada), demonstrating superiority of TREMFYA to U.S. licensed adalimumab.
An improvement was seen in psoriasis involving the scalp in subjects randomized to TREMFYA compared to placebo at Week 16.
Examination of age, gender, race, body weight, and previous treatment with systemic or biologic agents did not identify differences in response to TREMFYA among these subgroups.
Maintenance and Durability of Response
To evaluate maintenance and durability of response (trial PsO2), subjects randomized to TREMFYA at Week 0 and who were PASI 90 responders at Week 28 were re-randomized to either continue treatment with TREMFYA every 8 weeks or be withdrawn from therapy (i.e., receive placebo).
At Week 48, 89% of subjects who continued on TREMFYA maintained PASI 90 compared to 37% of subjects who were re-randomized to placebo and withdrawn from TREMFYA. For responders at Week 28 who were re-randomized to placebo and withdrawn from TREMFYA, the median time to loss of PASI 90 was approximately 15 weeks.
Patient Reported Outcomes
Greater improvements in symptoms of psoriasis (itch, pain, stinging, burning and skin tightness) at Week 16 in TREMFYA compared to placebo were observed in both trials based on the Psoriasis Symptoms and Signs Diary (PSSD). Greater proportions of subjects on TREMFYA compared to U.S. licensed adalimumab achieved a PSSD symptom score of 0 (symptom-free) at Week 24 in both trials.
Trial PsO3
Trial PsO3 [NCT02203032] evaluated the efficacy of 24 weeks of treatment with TREMFYA in subjects (N=268) who had not achieved an adequate response, defined as IGA ≥2 at Week 16 after initial treatment with U.S. licensed ustekinumab (dosed 45 mg or 90 mg according to the subject's baseline weight at Week 0 and Week 4). These subjects were randomized to either continue with U.S. licensed ustekinumab treatment every 12 weeks or switch to TREMFYA 100 mg at Weeks 16, 20, and every 8 weeks thereafter. Baseline characteristics for randomized subjects were similar to those observed in PsO1 and PsO2.
In subjects with an inadequate response (IGA ≥2 at Week 16 to U.S. licensed ustekinumab), greater proportions of subjects on TREMFYA compared to U.S. licensed ustekinumab achieved an IGA score of 0 or 1 with a ≥2 grade improvement at Week 28 (31% vs. 14%, respectively; 12 weeks after randomization).
Trial PsO4
Trial PsO4 [NCT02905331] evaluated the efficacy, safety, and pharmacokinetics of TREMFYA administered with the One-Press injector. In this trial, 78 subjects were randomized to receive either TREMFYA (100 mg at Weeks 0 and 4 and every 8 weeks thereafter) [N=62], or placebo [N=16]. Baseline characteristics for subjects were comparable to those observed in trials PsO1 and PsO2. The co-primary endpoints were the same as those for trials PsO1 and PsO2. Secondary endpoints included the proportion of subjects who achieved an IGA score of 0 at Week 16 and the proportion of subjects who achieved a PASI 100 response at Week 16.
A greater proportion of subjects in the guselkumab group achieved an IGA score of 0 or 1 or a PASI 90 response at Week 16 (81% and 76%, respectively) than in the placebo group (0% for both endpoints). The proportion of subjects who achieved an IGA score of 0 at Week 16 was higher in the guselkumab group compared to the placebo group (56% vs. 0%). The proportion of subjects who achieved a PASI 100 response at Week 16 was higher in the guselkumab group compared to the placebo group (50% vs. 0%).
Pediatric Subjects with Moderate-to-Severe Plaque Psoriasis
Trial PsO5
One multicenter, randomized, placebo and active biological comparator-controlled trial (PsO5 [NCT03451851]) enrolled 120 subjects 6 years to 17 years of age with moderate-to-severe plaque psoriasis who were candidates for systemic therapy or phototherapy and inadequately controlled by phototherapy and/or topical therapies.
In trial PsO5, 92 subjects were randomized to receive subcutaneous injection of either TREMFYA (N=41) or placebo (N=25) at Weeks 0, 4, and 12 or an active biological comparator (N=26). An additional 28 subjects enrolled in a TREMFYA open-label arm. Subjects with a body weight of 70 kg or more received 100 mg of TREMFYA. Subjects with a body weight less than 70 kg received doses according to their weight.
Subjects had an IGA score of ≥3 ("moderate") on a 5-point scale of overall disease severity, a PASI ≥12, and a minimum affected BSA of ≥10%, and at least one of the following: 1) very thick lesions, 2) clinically relevant facial, genital, or hand/foot involvement, 3) PASI ≥20, 4) BSA >20%, or 5) IGA=4 ("severe"). Subjects with guttate, erythrodermic, or pustular psoriasis were excluded.
Of the 92 subjects in the controlled part of the trial, 55% of subjects were male, 85% were White, 4% were Asian, and 4% were Black or African American. For ethnicity, 95% of subjects identified as Not Hispanic or Latino and 5% of subjects identified as Hispanic or Latino. Subjects had a mean body weight of 57 kg, a mean age of 13 years, and a third of the subjects were less than 12 years of age. At baseline, subjects had a median affected BSA of 20%, a median PASI score of 17, and 3% had a history of psoriatic arthritis. Approximately 22% of subjects had an IGA score of 4 ("severe"). Prior phototherapy or prior conventional systemic therapy was received by 32% of subjects and prior biologic systemic therapy was received by 10% of subjects.
The responses at Week 16 were compared to placebo for the two co-primary endpoints:
- the proportion of subjects who achieved an IGA score of 0 ("cleared") or 1 ("minimal");
- the proportion of subjects who achieved a PASI 90 response.
Other evaluated outcomes included the proportion of subjects who achieved PASI 75, IGA score of 0 ("cleared") and PASI 100 at Week 16.
Clinical Response
Table 7 presents the efficacy results at Week 16 in trial PsO5.
14.2 Clinical Studies in Adults with Psoriatic Arthritis
The safety and efficacy of TREMFYA were assessed in 2 randomized, double-blind, placebo-controlled trials (PsA1 [NCT03162796] and PsA2 [NCT03158285]) in 1120 adult subjects with active psoriatic arthritis (PsA) (≥3 swollen joints, ≥3 tender joints, and a C-reactive protein (CRP) level of ≥0.3 mg/dL in PsA1 and ≥5 swollen joints, ≥5 tender joints, and a CRP level of ≥0.6 mg/dL in PsA2) who had inadequate response to standard therapies (e.g., conventional DMARDs [cDMARDs]), apremilast, or nonsteroidal anti-inflammatory drugs [NSAIDs]). Subjects in these trials had a diagnosis of PsA for at least 6 months based on the Classification criteria for Psoriatic Arthritis (CASPAR) and a median duration of PsA of 4 years at baseline.
In trial PsA1, approximately 31% of subjects had been previously treated with up to 2 anti-tumor necrosis factor alpha (anti-TNFα) agents whereas in trial PsA2, all subjects were biologic naïve. Approximately 58% of subjects from both trials had concomitant methotrexate (MTX) use. Subjects with different subtypes of PsA were enrolled in both trials, including polyarticular arthritis with the absence of rheumatoid nodules (40%), spondylitis with peripheral arthritis (30%), asymmetric peripheral arthritis (23%), distal interphalangeal involvement (7%) and arthritis mutilans (1%). At baseline, over 65% and 42% of the subjects had enthesitis and dactylitis, respectively and 79% had ≥3% body surface area (BSA) psoriasis skin involvement.
Trial PsA1 evaluated 381 subjects who were treated with placebo SC, TREMFYA 100 mg SC at Weeks 0, 4 and every 8 weeks (q8w) thereafter, or TREMFYA 100 mg SC every 4 weeks (q4w). Trial PsA2 evaluated 739 subjects who were treated with placebo SC, TREMFYA 100 mg SC at Weeks 0, 4 and q8w thereafter, or TREMFYA 100 mg SC q4w. The primary endpoint in both trials was the percentage of subjects achieving an ACR20 response at Week 24.
Clinical Response
In both trials, subjects treated with TREMFYA 100 mg q8w demonstrated a greater clinical response including ACR20, compared to placebo at Week 24 (Tables 8 and 9). Similar responses were seen regardless of prior anti-TNFα exposure in PsA1, and in both trials similar responses were seen regardless of concomitant cDMARD use, previous treatment with cDMARDs, gender and body weight.
The percentage of subjects achieving ACR20 response in PsA2 by visit is shown in Figure 1.
Figure 1: Percentage of Adult Subjects with Active Psoriatic Arthritis Achieving ACR 20 Response by Visit Through Week 24 in Trial PsA2

The results of the components of the ACR response criteria are shown in Table 10.
Treatment with TREMFYA resulted in an improvement in the skin manifestations of psoriasis in subjects with PsA.
Treatment with TREMFYA resulted in improvement in dactylitis and enthesitis in subjects with pre-existing dactylitis or enthesitis.
Physical Function
TREMFYA treated subjects in the TREMFYA 100 mg q8w group in both trials PsA1 and PsA2 showed greater mean improvement from baseline in physical function compared to subjects treated with placebo as assessed by the Health Assessment Questionnaire-Disability Index (HAQ-DI) at Weeks 16 and 24. In both trials, the proportion of HAQ-DI responders (≥0.35 improvement in HAQ-DI score) was greater in the TREMFYA q8w dose group compared to placebo at Weeks 16 and 24.
Other Health-Related Outcomes
General health status was assessed by the Short Form health survey (SF-36). At Week 24, subjects in the TREMFYA 100 mg q8w dose group in both trials PsA1 and PsA2 showed greater improvement from baseline in the SF-36 physical component summary (PCS) compared with placebo. There was not a statistically significant improvement observed in the SF-36 MCS. At Week 24, there was numerical improvement in the physical functioning, role-physical, bodily-pain, general health, social-functioning and vitality domains but not in the role-emotional and mental health domains. Fatigue was assessed by the Functional Assessment of Chronic Illness Therapy-Fatigue score (FACIT-F) in trials PsA1 and PsA2. Treatment with TREMFYA resulted in improvement in fatigue as measured by FACIT-F.
14.3 Clinical Studies in Adults with Ulcerative Colitis
The efficacy and safety of TREMFYA was assessed in three randomized, double-blind, placebo-controlled trials (UC1, UC2, and UC 4) that enrolled adult subjects with moderately to severely active ulcerative colitis. Disease activity was assessed by the modified Mayo score (mMS), a 3-component Mayo score (0 –9) which consists of the following subscores (0 to 3 for each subscore): stool frequency (SFS), rectal bleeding (RBS), and findings on centrally reviewed endoscopy (ES). An ES of 2 was defined by marked erythema, lack of vascular pattern, friability, and/or erosions; an ES of 3 was defined by spontaneous bleeding and ulceration. Enrolled subjects with a mMS between 5 and 9 and an ES of 2 or 3 were classified as having moderately to severely active ulcerative colitis. Subjects with inadequate response, loss of response, or intolerance to corticosteroids, immunomodulators (azathioprine, 6-mercaptopurine), biologic therapy (TNF blockers, vedolizumab), and/or Janus kinase (JAK) inhibitors were enrolled. In UC4, subjects with inadequate response, loss of response, or intolerance to sphingosine-1-phosphate receptor modulators (S1PRM) were also enrolled.
Induction Trial: UC1
In the 12-week induction trial (UC1; NCT04033445), 701 adult subjects with moderately to severely active ulcerative colitis were randomized 3:2 to receive either TREMFYA 200 mg or placebo by intravenous infusion at Week 0, Week 4, and Week 8.
At baseline in trial UC1, the median mMS was 7, 64% of subjects had severely active disease (mMS ≥7), and 68% of subjects had an ES of 3. In trial UC1, 49% of subjects had previously failed (inadequate response, loss of response, or intolerance) treatment with at least one biologic therapy and/or JAK inhibitor, 48% were biologic and JAK inhibitor naïve, and 3% had previously received but not failed a biologic or JAK inhibitor. The median age was 39 years (ranging from 18 to 79 years); 43% were female; and 72% identified as White, 21% as Asian, 1% as Black or African American, <1% as American Indian or Alaskan Native, and <1% as multiple racial groups.
Enrolled subjects were permitted to use stable doses of oral aminosalicylates, immunomodulators (azathioprine, 6-mercaptopurine, methotrexate), and/or oral corticosteroids (up to 20 mg/day prednisone or equivalent). At baseline, 72% of subjects were receiving aminosalicylates, 21% of subjects were receiving immunomodulators, and 43% of subjects were receiving corticosteroids. Concomitant biologic therapies or JAK inhibitors were not permitted.
In trial UC1, the primary endpoint was clinical remission at Week 12 as defined by the mMS. Secondary endpoints at Week 12 included endoscopic improvement, clinical response, and histologic endoscopic mucosal improvement (see Table 11).
Trial UC1 was not designed to evaluate the relationship of histologic endoscopic mucosal improvement at Week 12 to disease progression and long-term outcomes.
Rectal Bleeding and Stool Frequency Subscores
Decreases in rectal bleeding and stool frequency subscores were observed as early as Week 4 in subjects treated with intravenous TREMFYA compared to placebo.
Endoscopic Assessment
Normalization of the endoscopic appearance of the mucosa (endoscopic remission) was defined as ES of 0. At Week 12 of UC1, a greater proportion of subjects treated with TREMFYA compared to placebo-treated subjects achieved endoscopic remission (15% vs 5%).
Fatigue Response
In trial UC1, subjects treated with TREMFYA experienced a clinically meaningful improvement in fatigue, assessed by the PROMIS-Fatigue Short form 7a, at Week 12, compared to placebo-treated subjects. The effect of TREMFYA to improve fatigue after 12 weeks of induction has not been established.
Maintenance Trial: UC2
The maintenance trial (UC2) evaluated 568 subjects who received one of two intravenous TREMFYA induction regimens, including the recommended 200 mg regimen, for 12 weeks in trials UC1 or UC3 (induction dose-finding study) and demonstrated clinical response per mMS after 12 weeks. Subjects were re-randomized to receive a subcutaneous maintenance regimen of either TREMFYA 100 mg every 8 weeks, TREMFYA 200 mg every 4 weeks, or placebo for up to an additional 44 weeks.
In trial UC2, 42% of subjects had failed (inadequate response, loss of response, or intolerance) treatment with one or more biologics or JAK inhibitors.
The primary endpoint was clinical remission at Week 44 defined by mMS. Secondary endpoints included corticosteroid-free clinical remission, endoscopic improvement, histologic endoscopic mucosal improvement, all at Week 44 and maintenance of clinical remission at Week 44 in subjects who achieved clinical remission 12 weeks after intravenous TREMFYA induction treatment (see Table 12).
Trial UC2 was not designed to evaluate the relationship of histologic endoscopic mucosal improvement at Week 44 to disease progression and long-term outcomes.
Endoscopic Assessment
Normalization of the endoscopic appearance of the mucosa (endoscopic remission) was defined as ES of 0. In UC2, greater proportions of subjects treated with TREMFYA 100 mg every 8 weeks or TREMFYA 200 mg every 4 weeks achieved endoscopic remission at Week 44 compared to placebo-treated subjects (35% and 34%, respectively, vs. 15%).
Trial UC4
In trial UC4 (NCT05528510), subjects with moderately to severely active ulcerative colitis were randomized 1:1:1 to receive TREMFYA 400 mg subcutaneous induction at Weeks 0, 4, and 8 followed by TREMFYA 200 mg subcutaneous maintenance every 4 weeks beginning at Week 12; TREMFYA 400 mg subcutaneous induction at Weeks 0, 4, and 8 followed by TREMFYA 100 mg subcutaneous maintenance every 8 weeks beginning at Week 16; or placebo. Efficacy was evaluated in 395 randomized subjects.
At baseline in trial UC4, the median mMS was 7; 65% of subjects had severely active disease (mMS ≥7), and 59% of subjects had an ES of 3. A total of 41% of subjects had previously failed treatment with at least one biologic therapy, JAK inhibitor, and/or S1PRM, 57% were biologic, JAK inhibitor, and S1PRM naïve, and 2% had previously received but had not failed a biologic, JAK inhibitor, or S1PRM. The median age of subjects was 40 years (ranging from 18 to 80 years); 39% were female; and 64% identified as White, 30% as Asian, and 3% as Black or African American.
Enrolled subjects were permitted to use stable doses of oral aminosalicylates, immunomodulators (azathioprine, 6-mercaptopurine, methotrexate), and/or oral corticosteroids (up to 20 mg/day prednisone or equivalent). At baseline, 78% of subjects were receiving aminosalicylates, 21% of subjects were receiving immunomodulators, and 33% of subjects were receiving corticosteroids. Concomitant biologic therapies, JAK inhibitors, or S1PRMs were not permitted.
In trial UC4, the primary endpoint was clinical remission at Week 12 as defined by the mMS. Secondary endpoints at Week 12 included endoscopic improvement, clinical response, and histologic-endoscopic mucosal improvement. Secondary endpoints at Week 24 included clinical remission and endoscopic improvement. The results of analyses of multiplicity-controlled efficacy endpoints in trial UC4 are shown in Table 13.
Rectal Bleeding and Stool Frequency Subscores
Decreases in rectal bleeding and stool frequency subscores were observed as early as Week 3 in subjects treated with subcutaneous TREMFYA compared to placebo.
Endoscopic Assessment
Normalization of the endoscopic appearance of the mucosa (endoscopic remission) was defined as ES of 0. At Week 12 of UC4, a greater proportion of subjects treated with TREMFYA compared to placebo-treated subjects achieved endoscopic remission (16% vs 2%). Greater proportions of subjects treated with TREMFYA 100 mg every 8 weeks or TREMFYA 200 mg every 4 weeks achieved endoscopic remission at Week 24 compared to placebo-treated subjects (20% and 27%, respectively, vs 3%).
14.4 Clinical Studies in Adults with Crohn's Disease
The efficacy and safety of TREMFYA were assessed in three randomized, double-blind, placebo-controlled trials that enrolled adult subjects with moderately to severely active Crohn's disease who had a history of inadequate response, loss of response, or intolerance to oral corticosteroids, immunomodulators (azathioprine, 6-mercaptopurine, methotrexate), and/or biologic therapy (TNF blockers or vedolizumab). Moderately to severely active Crohn's disease was defined as a Crohn's Disease Activity Index (CDAI) score of ≥220 and a Simple Endoscopic Score for Crohn's Disease (SES-CD) of ≥6 (or ≥4 for subjects with isolated ileal disease). Subjects were permitted to use stable doses of oral corticosteroids (prednisone ≤40 mg/day or equivalent), immunomodulators (azathioprine, 6-mercaptopurine, methotrexate), and/or aminosalicylates.
Trials CD1 and CD2
In trial CD1 (NCT03466411), 361 subjects were randomized to receive intravenous TREMFYA 200 mg at Weeks 0, 4, and 8 (N = 285) or placebo (N = 76). The median age of subjects enrolled into trial CD1 was 33 years (range: 18 – 83 years); 46% were female; and 75% identified as White, 22% as Asian, 1% as Black or African American, <1% as Native Hawaiian or Pacific Islander, and 2% did not report their racial group. The median baseline CDAI score was 285 (range: 220 – 442), and the median baseline SES-CD score was 11 (range: 4 – 39). Of the randomized subjects, 52% of subjects had previously failed (inadequate response, loss of response, or intolerance) treatment with at least one biologic therapy, 43% were biologic-naïve, and 6% had previously received but had not failed a biologic. At baseline, 37% of subjects were receiving oral corticosteroids and 30% of subjects were receiving immunomodulators (azathioprine, 6-mercaptopurine, methotrexate).
In trial CD2 (NCT03466411), 360 subjects were randomized to receive intravenous TREMFYA 200 mg at Weeks 0, 4, and 8 (N = 288) or placebo (N = 72). The median age of subjects enrolled into trial CD2 was 33 years (range:18 – 72 years); 39% were female; and 73% identified as White, 23% as Asian, 1% as Black or African American, <1% as Native Hawaiian or Pacific Islander, and 2% did not report their racial group. The median baseline CDAI score was 286 (range: 220 – 442) and the median baseline SES-CD score was 11 (range: 4 – 42). Of the randomized subjects, 52% of subjects had previously failed (inadequate response, loss of response, or intolerance) treatment with at least one biologic therapy, 41% were biologic-naïve, and 7% had previously received but had not failed a biologic. At baseline, 36% of the subjects were receiving oral corticosteroids and 31% of the subjects were receiving immunomodulators (azathioprine, 6-mercaptopurine, methotrexate).
The results of efficacy endpoints at Week 12 for CD1 and CD2 trials are shown in Table 14.
Stool Frequency and Abdominal Pain
Greater reductions in stool frequency and abdominal pain were observed as early as Week 4 in subjects treated with intravenous TREMFYA compared to placebo.
Trial CD3
In trial CD3, 340 subjects were randomized in a 1:1:1 ratio to receive subcutaneous TREMFYA 400 mg at Weeks 0, 4, and 8 followed by subcutaneous TREMFYA 100 mg every 8 weeks (with the first dose given at Week 16); subcutaneous TREMFYA 400 mg at Weeks 0, 4, and 8 followed by subcutaneous TREMFYA 200 mg every 4 weeks (with the first dose given at Week 12); or placebo. The median age of subjects enrolled into trial CD3 was 36 years (range: 18 – 83 years); 41% were female; and 66% identified as White, 22% as Asian, 3% as Black or African American, and 9% did not report their racial group. The median baseline CDAI score was 291 (range: 220 – 447), and the median baseline SES-CD score was 10 (range: 4 – 40). Of the randomized subjects, 46% of subjects had previously failed (inadequate response, loss of response, or intolerance) treatment with at least one biologic therapy, 47% were biologic naïve, and 7% had previously received but had not failed a biologic. At baseline, 30% of the subjects were receiving oral corticosteroids and 29% of the subjects were receiving immunomodulators (azathioprine, 6-mercaptopurine, methotrexate).
In trial CD3, the coprimary endpoints were clinical remission at Week 12 and endoscopic response at Week 12 compared to placebo. Additional efficacy endpoints included clinical response at Week 12, clinical remission at Week 24, clinical remission at Week 48, and endoscopic response at Week 48. The results of analyses of multiplicity-controlled efficacy endpoints in trial CD3 are shown in Table 15.
Stool Frequency and Abdominal Pain
Greater reductions in stool frequency and abdominal pain were observed as early as Week 4 in subjects treated with subcutaneous TREMFYA 400 mg compared to placebo.
Endoscopic Remission at Week 48
Endoscopic remission was defined as an SES-CD score ≤4 and at least a 2-point reduction from baseline and no subscore greater than 1 in any individual component. In trial CD3, a greater proportion of subjects treated with either TREMFYA regimen (i.e., subcutaneous TREMFYA 400 mg at Weeks 0, 4, and 8 followed by subcutaneous TREMFYA 100 mg at Week 16 and every 8 weeks thereafter or subcutaneous TREMFYA 400 mg at Weeks 0, 4, and 8 followed by subcutaneous TREMFYA 200 mg at Week 12 and every 4 weeks thereafter) achieved endoscopic remission, compared to placebo-treated subjects (31% and 40%, respectively, vs. 6%).
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
TREMFYA® (guselkumab) injection is a clear and colorless to light yellow solution supplied as follows:
Subcutaneous Injection
- Carton of one 100 mg/mL single-dose One-Press patient‑controlled injector (NDC: 57894-640-11)
- Carton of one 100 mg/mL single-dose prefilled pen (TREMFYA PEN) (NDC: 57894-640-06)
- Carton of one 200 mg/2 mL single-dose prefilled pen (TREMFYA PEN) (NDC: 57894-651-02)
- Induction Pack for Ulcerative Colitis or Crohn's Disease: Carton containing two 200 mg/2 mL single-dose prefilled pens (400 mg/4 mL total) (TREMFYA PEN) (NDC: 57894-651-04)
- Carton of one 100 mg/mL single-dose prefilled syringe with a 27G, half inch fixed needle assembled in a passive needle guard delivery system (NDC: 57894-640-01)
- Carton of one 200 mg/2 mL (100 mg/mL) single-dose prefilled syringe with a 27G, half inch fixed needle assembled in a passive needle guard delivery system (NDC: 57894-651-22)
Intravenous Infusion
- Carton of one 200 mg/20 mL (10 mg/mL) single-dose vial (NDC: 57894-650-02)
Storage and Handling
TREMFYA is sterile and preservative-free. Discard any unused portion.
- Store in a refrigerator between 2 °C to 8 °C (36 °F to 46 °F).
- Store in original carton until time of use.
- Protect from light until use.
- Do not freeze.
- Do not shake.
- Not made with natural rubber latex.
17 PATIENT COUNSELING INFORMATION
Advise the patients and/or caregivers to read the FDA-approved patient labeling (Medication Guide and Instructions for Use) before starting TREMFYA therapy, and each time the prescription is renewed, as there may be new information they need to know.
Hypersensitivity Reactions
Advise patients to discontinue TREMFYA and seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions [see Warnings and Precautions (5.1)].
Infections
Instruct patients of the importance of communicating any history of infections to the healthcare provider and contacting their healthcare provider if they develop any symptoms of an infection [see Warnings and Precautions (5.2)].
Tuberculosis
Advise patients to contact their healthcare provider if they experience symptoms suggestive of TB (e.g., unexplained fever, cough, or difficulty breathing) [see Warnings and Precautions (5.3)].
Hepatotoxicity
Inform patients that TREMFYA may cause liver injury. Instruct patients to seek immediate medical attention if they experience symptoms suggestive of liver dysfunction (e.g., unexplained rash, nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine) [see Warnings and Precautions (5.4)].
Immunizations
Advise patients treated with TREMFYA to avoid use of live vaccines [see Warnings and Precautions (5.5)].
Ulcerative Colitis and Crohn's Disease Subcutaneous Induction Dosing Instructions (400 mg Subcutaneous dose)
If patients are to receive the 400 mg subcutaneous induction dosage for ulcerative colitis or Crohn's disease, instruct patients or caregivers to administer two 200 mg prefilled pens or prefilled syringes to achieve the full 400 mg dose [see Medication Guide and Instructions for Use].
Instruction on Injection Technique
Instruct patients or caregivers to perform the first self-injection under the supervision and guidance of a qualified healthcare professional for proper training in subcutaneous injection technique, including injection of the full dose [see Medication Guide and Instructions for Use].
Proper Sharps Disposal Instructions
Counsel patients and caregivers on the proper technique of needle and syringe disposal. Instruct patients to dispose needles and syringes in a puncture-resistant container. Advise patients and caregivers not to reuse needles or syringes.
Administration Instructions
Instruct patients that if they forget to administer a dose of TREMFYA, then they should administer TREMFYA as soon as they remember. Instruct patients to administer the next dose at the regular scheduled time.
Pregnancy
Advise patients that there is a pregnancy registry that monitors pregnancy outcomes in patients exposed to TREMFYA during pregnancy [see Use in Specific Populations (8.1)].
Manufactured by:
Janssen Biotech, Inc.
Horsham, PA 19044, USA
U.S. License No. 1864
For patent information: www.janssenpatents.com
© Johnson & Johnson and its affiliates 2017–2025
INSTRUCTIONS FOR USE
TREMFYA® [trem fye' ah]
(guselkumab)
injection, for subcutaneous use
200 mg/2mL (100 mg/mL) Prefilled Syringe
This Instructions for Use contains information on how to inject TREMFYA.

Important Information You Need to Know Before Injecting TREMFYA
TREMFYA comes in a single-dose prefilled syringe containing one 200 mg dose.
If your healthcare provider decides that you or a caregiver may be able to give your injections of TREMFYA at home, you should receive training on the correct way to prepare and inject TREMFYA before using the prefilled syringe.
Read this Instructions for Use before using your TREMFYA prefilled syringe and each time you get a refill. There may be new information. This leaflet does not take the place of talking with your healthcare provider about your medical condition or your treatment.
Each TREMFYA prefilled syringe can only be used one time. Throw the used prefilled syringe away (see Step 4) after one dose, even if there is still medicine left in it. Do not reuse your TREMFYA prefilled syringe.
The TREMFYA prefilled syringe is intended for injection under the skin, not into the muscle or vein. After injection, the needle will retract into the device and lock into place.

Store in refrigerator between 36°F to 46°F (2°C to 8°C).
Do not freeze TREMFYA prefilled syringe.
Do not shake your TREMFYA prefilled syringe.
Keep TREMFYA prefilled syringe and all medicines out of reach of children.
Keep TREMFYA prefilled syringe in the original carton to protect from light and physical damage.
Prefilled syringe parts

1. Get ready

Check your dose to see if you will need to use 1 or 2 prefilled syringes and inspect carton(s)
Remove the carton(s) from the refrigerator.
Check the expiration ('EXP') date. Do not use your prefilled syringe if the expiration date has passed or if the seal on the carton is broken. Contact your healthcare provider or pharmacist for a new prefilled syringe.

Allow TREMFYA to come to room temperature
Let the cartons sit on a flat surface at room temperature for approximately 30 minutes before use.
Do not warm the prefilled syringe(s) any other way.
2. Prepare to inject TREMFYA

Take the prefilled syringe out of the carton.
Inspect liquid to see that it is clear and colorless to slightly yellow
Check the liquid in the viewing window. It should be clear and colorless to slightly yellow and may contain tiny white or clear particles. You may also see air bubbles. This is normal.
Do not inject if the liquid is:
- cloudy or
- discolored or
- has large particles
Do not use the prefilled syringe if it is dropped. Call your healthcare provider or pharmacist for a new prefilled syringe.

Choose injection site
Select a site from the following areas for your injection:
- Front of thighs
- Lower stomach area (lower abdomen), except for a 2-inch area right around your navel (belly-button)
- Back of upper arms (only if someone else is giving you the injection)
If you need to give 2 injections to complete your dose, choose different areas or leave at least 2 inches between injection sites.
Do not inject into skin that is tender, bruised, red, scaly, thick or hard. Avoid areas with scars or stretch marks.

Wash hands and clean injection site
Wash your hands well with soap and warm water.
Wipe your chosen injection site with an alcohol swab and allow it to dry.
Do not touch, fan, or blow on the injection site after you have cleaned it.
3. Inject TREMFYA

Remove needle cover when you are ready to inject
Hold the prefilled syringe by the body and pull needle cover straight off. It is normal to see a few drops of liquid.
Inject TREMFYA within 5 minutes of removing the needle cover.
Do not put needle cover back on, as this may damage the needle or cause a needle stick injury.
Do not touch needle or let it touch any surface.
Do not use the prefilled syringe if it is dropped. Call your healthcare provider or pharmacist for a new prefilled syringe.
Do not hold or pull the plunger at any time.

Pinch injection site and insert needle at about a 45-degree angle
It is important to pinch enough skin to inject under the skin and not into muscle.
Insert needle with a quick dart-like motion.

Slowly press plunger all the way down until it stops to inject all of the liquid
You will feel some resistance as you press the plunger, this is normal.

Release pressure from plunger to remove the needle from the skin
The needle will retract into the device and lock into place.
If your prescribed dose requires two injections, repeat Steps 2 to 4 with the second prefilled syringe.
4. After your injection

Check injection site
There may be a small amount of blood or liquid at the injection site. Gently hold pressure over the injection site with a cotton ball or gauze pad until any bleeding stops.
Do not rub the injection site.
If needed, cover the injection site with a bandage.

Dispose of your prefilled syringe
Put the used prefilled syringe in an FDA-cleared sharps disposal container right away after use.
Do not throw away (dispose of) your prefilled syringe in your household trash.
Do not recycle your used sharps disposal container.
For more information, see Disposing of TREMFYA prefilled syringe.
Disposing of TREMFYA prefilled syringe
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic
- can be closed with a tight-fitting, puncture resistant lid, without sharps being able to come out
- upright and stable during use
- leak-resistant
- properly labeled to warn of hazardous waste inside the container
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: www.fda.gov/safesharpsdisposal. You may also consult your pharmacist.

Call your healthcare provider to talk about any questions you may have. For additional assistance or to share your feedback, call 800-526-7736.
Manufactured by:
Janssen Biotech, Inc.
Horsham, PA 19044, USA
US License No. 1864
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Approved: August / 2025
INSTRUCTIONS FOR USE
TREMFYA® PEN
[trem fye' ah Pen]
(guselkumab)
injection, for subcutaneous use
200 mg/2 mL Prefilled Pen
This Instructions for Use contains information on how to inject with TREMFYA PEN.

Important Information You Need to Know Before Injecting with TREMFYA PEN
TREMFYA PEN comes in a single-dose Prefilled Pen containing one 200 mg dose.
If your healthcare provider decides that you or a caregiver may be able to give your injections with TREMFYA PEN at home, you should receive training on the correct way to prepare and inject with TREMFYA PEN before using the Prefilled Pen.
Read this Instructions for Use before using your Prefilled Pen and each time you get a new Prefilled Pen. There may be new information. This leaflet does not take the place of talking with your healthcare provider about your medical condition or your treatment.
Each TREMFYA PEN can only be used one time. Throw the used Prefilled Pen away (see Step 4) after one dose, even if there is still medicine left in it. Do not reuse your Prefilled Pen.

Store in refrigerator between 36°F to 46°F (2°C to 8°C).
Do not freeze your TREMFYA PEN.
Do not shake your TREMFYA PEN.
Keep your TREMFYA PEN and all medicines out of reach of children.
Keep your TREMFYA PEN in the original carton to protect from light and physical damage.
TREMFYA PEN parts

1. Get ready

Check your prescribed dose to see if you will need to use 1 or 2 Prefilled Pens and inspect carton(s)
Remove the carton(s) from the refrigerator.
Check the expiration ('EXP') date.
Do not use the Prefilled Pen if the expiration date has passed or if the seal on the carton is broken. Contact your healthcare provider or pharmacist for a new Prefilled Pen.

Allow TREMFYA PEN to come to room temperature
Let the carton(s) sit on a flat surface at room temperature for approximately 30 minutes before use.
Do not warm the Prefilled Pen(s) any other way.
2. Prepare to inject with TREMFYA PEN

Take your Prefilled Pen out of the carton.
Inspect liquid in window to see that it is clear and colorless to slightly yellow
Check the liquid in the viewing window. It should be clear and colorless to slightly yellow and may contain tiny white or clear particles. You may also see air bubbles. This is normal.
Do not inject if the liquid is:
- cloudy or
- discolored or
- has large particles
Call your healthcare provider or pharmacist for a new Prefilled Pen.

Choose injection site
Select from the following areas for your injection:
- Front of thighs
- Lower stomach area (lower abdomen), except for a 2-inch area right around your navel (belly-button)
- Back of upper arms (only if someone else is giving you the injection)
If you need to give 2 injections to complete your dose, choose different areas or leave at least 2 inches between injection sites.
Do not inject into skin that is tender, bruised, red, scaly, thick or hard. Avoid areas with scars or stretch marks.

Wash hands and clean injection site
Wash your hands well with soap and warm water.
Wipe your chosen injection site with an alcohol swab and allow it to dry.
Do not touch, fan, or blow on the injection site after you have cleaned it.
3. Inject with TREMFYA PEN

Remove cap when you are ready to inject
Pull the cap straight off. It is normal to see a few drops of liquid.
Inject with TREMFYA PEN within 5 minutes of removing cap.
Do not put the cap back on as this may damage the needle.
Do not use your Prefilled Pen if it is dropped after removing the cap. Call your healthcare provider or pharmacist for a new Prefilled Pen.

Position the Prefilled Pen straight onto the injection site then push and hold the Prefilled Pen
Position your Prefilled Pen straight onto the injection site with the yellow needle guard against the skin and the viewing window facing you.
Press down on the Prefilled Pen and keep holding it down against the skin.
You will hear the first click.

Keep holding your Prefilled Pen firmly against the skin for about 10 seconds to hear a second click
You are almost done.

Keep holding firmly against the skin and confirm your injection is complete
Your injection is complete when the plunger rod stops moving and fills the viewing window.

Lift straight up
If your prescribed dose requires two injections, repeat Steps 2 to 4 with the second Prefilled Pen.
4. After your injection

Check injection site
There may be a small amount of blood or liquid at the injection site. Gently hold pressure over the injection site with a cotton ball or gauze pad until any bleeding stops.
Do not rub the injection site. If needed, cover the injection site with a bandage.

Dispose of your Prefilled Pen and cap
Put your used Prefilled Pen and cap in an FDA-cleared sharps disposal container right away after use.
Do not throw away (dispose of) your Prefilled Pen in your household trash.
Do not recycle your used sharps disposal container.
For more information, see Disposing of TREMFYA PEN.
Disposing of TREMFYA PEN
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic
- can be closed with a tight-fitting, puncture resistant lid, without sharps being able to come out
- upright and stable during use
- leak-resistant
- properly labeled to warn of hazardous waste inside the container
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: www.fda.gov/safesharpsdisposal. You may also consult your pharmacist.

Call your healthcare provider to talk about any questions you may have. For additional assistance or to share your feedback, call 800-526-7736.
Manufactured by:
Janssen Biotech, Inc.
Horsham, PA 19044, USA
US License No. 1864
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Approved: March / 2025
Instructions for Use
TREMFYA®
(trem fye' ah)
(guselkumab)
Prefilled Syringe

SINGLE-DOSE
Important
TREMFYA comes as a single-dose prefilled syringe containing one 100 mg dose. Each TREMFYA prefilled syringe can only be used one time. Throw the used prefilled syringe away (See Step 3) after one dose, even if there is medicine left in it. Do not reuse your TREMFYA prefilled syringe.
If your healthcare provider decides that you or a caregiver may be able to give your injections of TREMFYA at home, you should receive training on the right way to prepare and inject TREMFYA using the prefilled syringe before attempting to inject. Do not try to inject yourself until you have been shown the right way to give the injections by your healthcare provider.
For children, TREMFYA should be administered by a healthcare provider or an adult caregiver who has been shown the right way to give the injections by your healthcare provider.
Read this Instructions for Use before using your TREMFYA prefilled syringe and each time you get a refill. There may be new information. This leaflet does not take the place of talking with your healthcare provider about your medical condition or your treatment.
The TREMFYA prefilled syringe is intended for injection under the skin, not into the muscle or vein. After injection, the needle will retract into the body of the device and lock into place.

Store in refrigerator at 36 °F to 46 °F (2 °C to 8 °C). Do not freeze TREMFYA prefilled syringe.
Keep TREMFYA prefilled syringe and all medicines out of reach of children.
Do not shake your TREMFYA prefilled syringe.
Keep TREMFYA prefilled syringe in the original carton to protect from light and physical damage.
Prefilled syringe parts
Before use

After use

1. Prepare for your injection

Inspect carton
Remove your TREMFYA prefilled syringe carton from the refrigerator.
Keep the prefilled syringe in the carton and let it sit on a flat surface at room temperature for at least 30 minutes before use.
Do not warm the prefilled syringe any other way.
Check the expiration date ('EXP') on the back panel of the carton.
Do not use your prefilled syringe if the expiration date has passed.
Do not inject TREMFYA if the perforations on the carton are broken. Call your healthcare provider or pharmacist for a refill.

Choose injection site
Select from the following areas for your injection:
- Front of thighs (recommended)
- Lower stomach area (lower abdomen), except for a 2-inch area right around your navel (belly-button)
- Back of upper arms (only if someone else is giving you the injection)
Do not inject into skin that is tender, bruised, red, hard, thick, scaly or affected by psoriasis.

Clean injection site
Wash your hands well with soap and warm water.
Wipe your chosen injection site with an alcohol swab and allow it to dry.
Do not touch, fan, or blow on the injection site after you have cleaned it.

Inspect liquid
Take your TREMFYA prefilled syringe out of the carton.
Check the TREMFYA prefilled syringe liquid in the viewing window. It should be clear to slightly yellow and may contain tiny white or clear particles. You may also see one or more air bubbles. This is normal.
Do not inject if the liquid is cloudy or discolored, or has large particles. Call your healthcare provider or pharmacist for a refill.
2. Inject TREMFYA using prefilled syringe

Remove needle cover
Hold your prefilled syringe by the body and pull needle cover straight off. It is normal to see a drop of liquid.
Inject TREMFYA within 5 minutes of removing the needle cover.
Do not put needle cover back on, as this may damage the needle or cause a needle stick injury.
Do not touch needle or let it touch any surface.
Do not use a TREMFYA prefilled syringe if it is dropped. Call your healthcare provider or pharmacist for a refill.

Position fingers and insert needle
Place your thumb, index and middle fingers directly under the finger flange, as shown.
Do not touch plunger or area above finger flange as this may cause the needle safety device to activate.
Use your other hand to pinch skin at the injection site. Position syringe at about a 45 degree angle to the skin.
It is important to pinch enough skin to inject under the skin and not into the muscle.
Insert needle with a quick, dart-like motion.

Release pinch and reposition hand
Use your free hand to grasp the body of the prefilled syringe.

Press plunger
Place thumb from the opposite hand on the plunger and press the plunger all the way down until it stops.

Release pressure from plunger
The safety guard will cover the needle and lock into place, removing the needle from your skin.
3. After your injection

Dispose of your prefilled syringe
Put your used TREMFYA prefilled syringe in an FDA-cleared sharps disposal container right away after use.
Do not throw away (dispose of) your TREMFYA prefilled syringe in your household trash.
Do not recycle your used sharps disposal container.
For more information, see "How should I dispose of the used prefilled syringe?"

Check injection site
There may be a small amount of blood or liquid at the injection site. Hold pressure over your skin with a cotton ball or gauze pad until any bleeding stops.
Do not rub the injection site.
If needed, cover injection site with a bandage.

Call your healthcare provider to talk about any questions you may have. For additional assistance or to share your feedback call 1-800-526-7736.
How should I dispose of the used prefilled syringe?
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out
- upright and stable during use
- leak-resistant
- properly labeled to warn of hazardous waste inside the container
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: www.fda.gov/safesharpsdisposal
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Janssen Biotech, Inc.
Horsham, PA 19044, USA
U.S. License No. 1864
Approved: September 2025
INSTRUCTIONS FOR USE
TREMFYA®
[trem fye´ ah]
(guselkumab)
injection, for subcutaneous use
One-Press Patient-Controlled Injector
This "Instructions for Use" contains information on how to inject TREMFYA.

Important Information You Need to Know Before Injecting TREMFYA
TREMFYA comes in a single-dose One-Press injector containing one 100 mg dose.
Each One-Press injector can only be used one time. Throw away (See Step 3) after one dose, even if there is medicine left in it. Do not reuse your One-Press injector.
If your healthcare provider decides that you or a caregiver may be able to give your injections of TREMFYA at home, you should receive training on the right way to prepare and inject TREMFYA using the One-Press injector. Do not try to inject yourself until you have been trained by your healthcare provider.
For children, TREMFYA should be administered by a healthcare provider or an adult caregiver who has been shown the right way to give the injections by your healthcare provider.
Please read this Instructions for Use before using your One-Press injector and each time you get a new One-Press injector. There may be new information.
This leaflet does not take the place of talking with your healthcare provider about your medical condition or your treatment.

Store in refrigerator at 36 °F to 46 °F (2 °C to 8 °C).
Do not freeze your One-Press injector.
Do not shake your One-Press injector.
Keep your One-Press injector and all medicines out of reach of children.
Keep your One-Press injector in the original carton to protect from light and physical damage.

Call your healthcare provider to talk about any questions you may have. For additional assistance or to share your feedback call 1-800-526-7736.
One-Press injector parts
Before use After use

1. Preparing to Inject TREMFYA

Inspect carton and allow TREMFYA to come to room temperature
Remove your One-Press injector carton from the refrigerator.
Keep your One-Press injector in the carton and let it sit on a flat surface at room temperature for at least 30 minutes before use.
Do not warm your One-Press injector any other way.

Check the expiration date ('EXP') on the carton
Do not use your One-Press injector if the expiration date has passed.
Do not inject TREMFYA if the seal on the carton is broken. Call your healthcare provider or pharmacist for a new One-Press injector.

Choose injection site
Select from the following areas for your injection:
- Front of thighs (recommended)
- Lower stomach area (lower abdomen), except for a 2-inch area right around your navel (belly-button)
- Back of upper arms (only for adults and only if someone else is giving you the injection). This injection site is not for use in children.
Do not inject into skin that is tender, bruised, red, hard, thick, scaly, or affected by psoriasis.

Wash hands
Wash your hands well with soap and warm water.
Clean injection site
Wipe your chosen injection site with an alcohol swab and allow it to dry.
Do not touch, fan, or blow on the injection site after you have cleaned it.

Inspect liquid in window
Take your One-Press injector out of the carton.
Check the liquid in the window. It should be clear to slightly yellow and may contain tiny white or clear particles. You may also see one or more air bubbles. This is normal.
Do not inject if the liquid is:
- cloudy,
- discolored, or
- has large particles.
Call your healthcare provider or pharmacist for a new One-Press injector.
2. Injecting TREMFYA

Pull off bottom cap
Keep hands away from the needle guard after the cap is removed. It is normal to see a few drops of liquid.
Inject TREMFYA within 5 minutes of removing the cap.
Do not put the cap back on. This could damage the needle.
Do not use a One-Press injector if it is dropped after removing the cap. Call your healthcare provider or pharmacist for a new One-Press injector.

Place straight on skin.
Push handle all the way down until teal body is not visible
You may hear a click when the injection begins. Keep pushing.
If you feel resistance, keep pushing. This is normal.
The medication injects as you push. Do this at a speed that is comfortable for you.

Confirm your injection is complete
Your injection is complete when:
- The teal body is not visible.
- You cannot press the handle down anymore.
- You may hear a click.

Lift straight up
The yellow band indicates that the needle guard is locked.
3. After your injection

Dispose of your One-Press injector
Put your used One-Press injector in a sharps disposal container right away after use.
Do not throw away (dispose of) your One-Press injector in your household trash.
Do not recycle your used sharps disposal container.
For more information, see "Disposing of TREMFYA One-Press injector".

Check injection site
There may be a small amount of blood or liquid at the injection site. Hold pressure over your skin with a cotton ball or gauze pad until any bleeding stops.
Do not rub the injection site.
If needed, cover injection site with a bandage.
Disposing of TREMFYA One-Press injector
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out
- upright and stable during use
- leak-resistant
- properly labeled to warn of hazardous waste inside the container
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: www.fda.gov/safesharpsdisposal. You may also consult your pharmacist.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Janssen Biotech, Inc.
Horsham, PA 19044, USA
U.S. License No. 1864
Revised: September 2025
INSTRUCTIONS FOR USE
TREMFYA® PEN
[trem fye' ah Pen]
(guselkumab)
injection, for subcutaneous use
100 mg/mL Prefilled Pen
This Instructions for Use contains information on how to inject with TREMFYA PEN.

Important Information You Need to Know Before Injecting with TREMFYA PEN
TREMFYA PEN comes in a single-dose Prefilled Pen containing one 100 mg dose.
If your healthcare provider decides that you or a caregiver may be able to give your injections with TREMFYA PEN at home, you should receive training on the correct way to prepare and inject with TREMFYA PEN before using the Prefilled Pen.
For children, TREMFYA PEN should be administered by a healthcare provider or an adult caregiver who has been shown the right way to give the injections by your healthcare provider.
Read this Instructions for Use before using your Prefilled Pen and each time you get a new Prefilled Pen. There may be new information. This leaflet does not take the place of talking with your healthcare provider about your medical condition or your treatment.
Each TREMFYA PEN can only be used one time. Throw the used Prefilled Pen away (see Step 4) after one dose, even if there is still medicine left in it. Do not reuse your Prefilled Pen.

Store in refrigerator between 36 °F to 46 °F (2 °C to 8 °C).
Do not freeze your TREMFYA PEN.
Do not shake your TREMFYA PEN.
Keep your TREMFYA PEN and all medicines out of reach of children.
Keep your TREMFYA PEN in the original carton to protect from light and physical damage.
TREMFYA PEN parts

1. Get ready

Allow TREMFYA PEN to come to room temperature and inspect carton
Remove the carton from the refrigerator and let the carton sit on a flat surface at room temperature for approximately 30 minutes before use.
Do not warm the Prefilled Pen any other way.
Check to see that you have the correct medicine and dose.
Check the expiration ('EXP') date.
Do not use the Prefilled Pen if the expiration date has passed or if the seal on the carton is broken. Contact your healthcare provider or pharmacist for a new Prefilled Pen.
2. Prepare to inject with TREMFYA PEN

Take your Prefilled Pen out of the carton.
Inspect liquid in window to see that it is clear and colorless to slightly yellow
Check the liquid in the viewing window. It should be clear and colorless to slightly yellow and may contain tiny white or clear particles. You may also see air bubbles. This is normal.
Do not inject if the liquid is:
- cloudy or
- discolored or
- has large particles
Call your healthcare provider or pharmacist for a new Prefilled Pen.
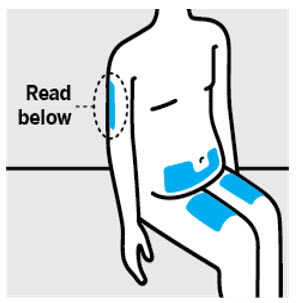
Choose injection site
Select from the following areas for your injection:
- Front of thighs
- Lower stomach area (lower abdomen), except for a 2-inch area right around your navel (belly-button)
- Back of upper arms (only for adults and only if someone else is giving you the injection). This injection site is not for use in children.
Do not inject into skin that is tender, bruised, red, scaly, thick or hard. Avoid areas with scars or stretch marks.
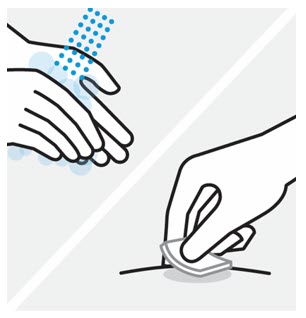
Wash hands and clean injection site
Wash your hands well with soap and warm water.
Wipe your chosen injection site with an alcohol swab and allow it to dry.
Do not touch, fan, or blow on the injection site after you have cleaned it.
3. Inject with TREMFYA PEN
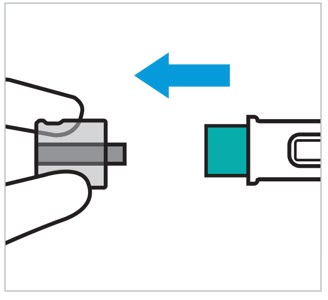
Remove cap when you are ready to inject
Pull the cap straight off. It is normal to see a few drops of liquid.
Inject with TREMFYA PEN within 5 minutes of removing cap.
Do not put the cap back on as this may damage the needle.
Do not use your Prefilled Pen if it is dropped after removing the cap. Call your healthcare provider or pharmacist for a new Prefilled Pen.
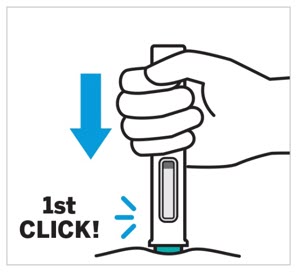
Position the Prefilled Pen straight onto the injection site then push and hold the Prefilled Pen
Position your Prefilled Pen straight onto the injection site with the teal needle guard against the skin and the viewing window facing you.
Press down on the Prefilled Pen and keep holding it down against the skin.
You will hear the first click.
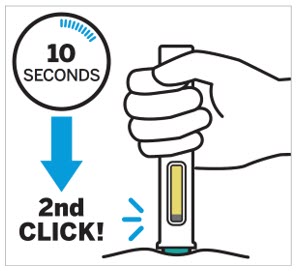
Keep holding your Prefilled Pen firmly against the skin for about 10 seconds to hear a second click
You are almost done.
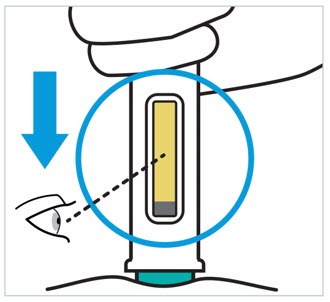
Keep holding firmly against the skin and confirm your injection is complete
Your injection is complete when the yellow plunger rod stops moving and fills the viewing window.
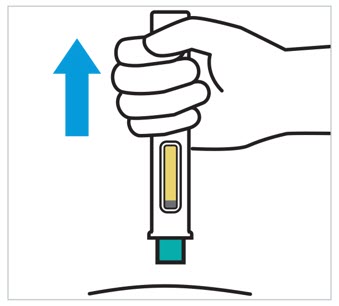
Lift straight up
4. After your injection
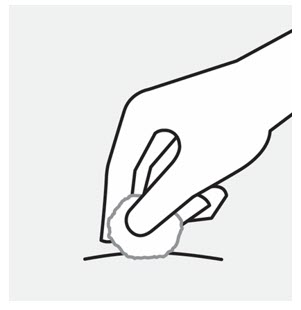
Check injection site
There may be a small amount of blood or liquid at the injection site. Gently hold pressure over the injection site with a cotton ball or gauze pad until any bleeding stops.
Do not rub the injection site. If needed, cover the injection site with a bandage.
Dispose of your Prefilled Pen and cap
Put your used Prefilled Pen and cap in an FDA-cleared sharps disposal container right away after use.
Do not throw away (dispose of) your Prefilled Pen in your household trash.
Do not recycle your used sharps disposal container.
For more information, see Disposing of TREMFYA PEN.
Disposing of TREMFYA PEN
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic
- can be closed with a tight-fitting, puncture resistant lid, without sharps being able to come out
- upright and stable during use
- leak-resistant
- properly labeled to warn of hazardous waste inside the container
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: www.fda.gov/safesharpsdisposal. You may also consult your pharmacist.

Call your healthcare provider to talk about any questions you may have. For additional assistance or to share your feedback, call 1-800-526-7736.
Manufactured by:
Janssen Biotech, Inc.
Horsham, PA 19044, USA
U.S. License No. 1864
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Approved: September / 2025
PRINCIPAL DISPLAY PANEL - 100 mg/mL Syringe Carton - NDC 57894-640-01
Tremfya®
(guselkumab)
Injection
100 mg/mL
FOR SUBCUTANEOUS USE ONLY
Rx only
NDC 57894-640-01
One single-dose prefilled syringe
Discard unused portion
Contains no preservative
ATTENTION: Dispense the enclosed
Medication Guide to each patient

PRINCIPAL DISPLAY PANEL - 100 mg/mL Syringe Carton - NDC 57894-640-11
Tremfya®
(guselkumab)
Injection
100 mg/mL
FOR SUBCUTANEOUS USE ONLY
Rx only
NDC 57894-640-11
Single-dose One-Press
patient-controlled injector
Discard unused portion
Contains no preservative
ATTENTION: Dispense the enclosed
Medication Guide to each patient

PRINCIPAL DISPLAY PANEL - 100 mg/mL Pen Carton
NDC 57894-640-06
Tremfya® PEN
(guselkumab)
Injection
100 mg/mL
FOR SUBCUTANEOUS USE ONLY
Rx only
One single-dose prefilled pen
Discard unused portion
Attention: Dispense the enclosed
Medication Guide to each patient.
PRINCIPAL DISPLAY PANEL - 200 mg/20 mL Vial Carton
NDC 57894-650-02
Tremfya®
(guselkumab)
Injection
200 mg/20 mL
(10 mg/mL)
FOR INTRAVENOUS
INFUSION ONLY
Attention: Dispense the
enclosed Medication Guide
to each patient.
Rx only
Single-dose vial
Discard unused portion
PRINCIPAL DISPLAY PANEL - 200 mg/2 mL Pen Carton
NDC 57894-651-02
Tremfya® PEN
(guselkumab)
Injection
200 mg/2 mL
FOR SUBCUTANEOUS USE ONLY
Rx only
One single-dose prefilled pen
Discard unused portion
ATTENTION: Dispense the enclosed
Medication Guide to each patient.
PRINCIPAL DISPLAY PANEL - 200 mg/2 mL Syringe Carton
NDC 57894-651-22
Tremfya®
(guselkumab)
Injection
200 mg/2 mL
FOR SUBCUTANEOUS USE ONLY
Rx only
One single-dose prefilled syringe
Discard unused portion
ATTENTION: Dispense the enclosed
Medication Guide to each patient.
PRINCIPAL DISPLAY PANEL - 200 mg/2 mL Pen Carton - NDC 57894-651-04
NDC 57894-651-04
Rx only
Tremfya® PEN
(guselkumab)
Injection
200 mg/2 mL
FOR SUBCUTANEOUS USE ONLY
Induction Pack for Crohn's Disease
and Ulcerative Colitis
(includes 1 induction dose)
The entire carton is to
be dispensed as a unit.
Contains: Two cartons each containing
one single-dose prefilled pen.
Discard unused portion.
ATTENTION: Dispense the enclosed
Medication Guide to each patient.

Guideline Central and select third party use “cookies” on this website to enhance the user experience.
This technology helps us gather statistical and analytical information to optimize the relevant content for you.
The user also has the option to opt-out which may have an effect on the browsing experience.