Sephience (sepiapterin) powder
1 INDICATIONS AND USAGE
SEPHIENCE is indicated for the treatment of hyperphenylalaninemia (HPA) in adult and pediatric patients 1 month of age and older with sepiapterin-responsive phenylketonuria (PKU). SEPHIENCE is to be used in conjunction with a phenylalanine (Phe)-restricted diet.
SEPHIENCE is a phenylalanine hydroxylase (PAH) activator indicated for the treatment of hyperphenylalaninemia (HPA) in adult and pediatric patients 1 month of age and older with sepiapterin-responsive phenylketonuria (PKU). SEPHIENCE is to be used in conjunction with a phenylalanine (Phe)- restricted diet. (1)
2 DOSAGE AND ADMINISTRATION
- Patients treated with SEPHIENCE should be on a dietary protein and a Phe-restricted diet. (2.1)
- Administer SEPHIENCE orally once daily with food. (2.2)
- The recommended starting dosage of SEPHIENCE is: (2.2)
- Important Administration Information: prepare SEPHIENCE calculated daily doses of < 1,000 mg as a liquid mixture (25 mg/mL), and administer exact prescribed dose volume (mL). (2.2, 2.3)
- See full prescribing information for complete preparation and administration instructions. (2.3)
2.1 Important Recommendation Prior to SEPHIENCE Treatment
Treatment with SEPHIENCE should be directed by physicians knowledgeable in the management of PKU.
Biochemical response to SEPHIENCE treatment cannot generally be pre-determined by laboratory testing (e.g., molecular testing), and should be determined through a therapeutic evaluation of SEPHIENCE [see Dosage and Administration (2.2) and Clinical Studies (14.1)].
Obtain baseline blood Phe concentration before initiating treatment.
All patients with PKU who are treated with SEPHIENCE should be on a dietary protein and Phe-restricted diet that is based on blood Phe levels. Patients should undergo regular dietary assessments, including protein and Phe intake, by their healthcare provider [see Dosage and Administration (2.2)].
2.2 Recommended Dosage and Administration
The recommended starting dosage of SEPHIENCE is based on the patient’s age and is administered orally once daily (see Table 1).
Administer SEPHIENCE with food [see Clinical Pharmacology (12.3)].
Evaluation Period
Dosage Titration in Patients Less than 2 Years of Age
After initiating treatment at the starting dosage by age (Table 1), check blood Phe levels to determine response to treatment within 2 weeks. If blood Phe does not decrease, SEPHIENCE dosage may be titrated incrementally based on blood Phe levels to a maximum daily dosage of 60 mg/kg. Existing dietary protein and Phe intake should not be modified during the evaluation period.
Discontinuation for Lack of Biochemical Response
Discontinue SEPHIENCE in patients whose blood Phe does not decrease after 2 weeks of treatment at the maximum daily dosage of 60 mg/kg.
Dosage Modification and Monitoring
Monitor blood Phe levels during treatment, and if needed, modify the daily dosage of SEPHIENCE within the range of 7.5 mg/kg to 60 mg/kg and/or dietary protein and Phe intake to ensure adequate blood Phe level control. Frequent blood Phe monitoring is recommended in the pediatric population [see Warnings and Precautions (5.2)].
Missed Dose
A missed dose should be taken as soon as possible but 2 doses should not be administered on the same day. Resume the normal dosing schedule the following day.
2.3 Preparation and Administration Instructions
Doses Less Than 1,000 mg (administration based on 25 mg/mL concentration)
- Determine the required number of SEPHIENCE packets and the required volume of water or apple juice to achieve a concentration of 25 mg/mL mixture (see Table 2).
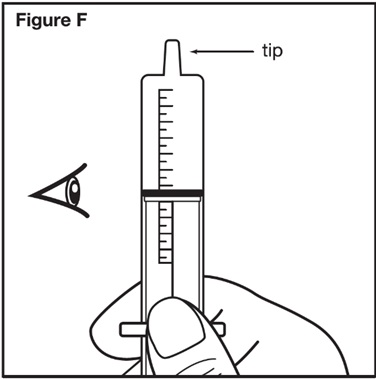
- Calculate the prescribed dose volume to the nearest 0.2 mL.
- Divide the calculated daily dose (mg) by the final concentration (25 mg/mL) of SEPHIENCE liquid mixture for doses less than 1,000 mg.
Prescribed dose volume (mL) = SEPHIENCE calculated dose (mg))
25 mg/mL
- Divide the calculated daily dose (mg) by the final concentration (25 mg/mL) of SEPHIENCE liquid mixture for doses less than 1,000 mg.
- Prepare a liquid mixture.
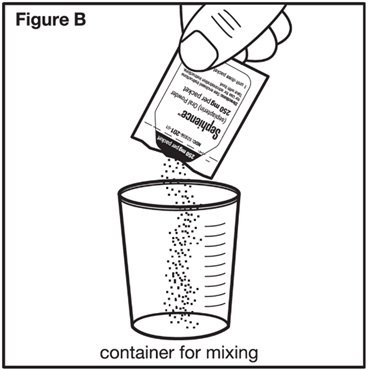
- Open and empty the entire content of each SEPHIENCE packet into an appropriate-size container and mix with the required volume of water or apple juice per Table 2.
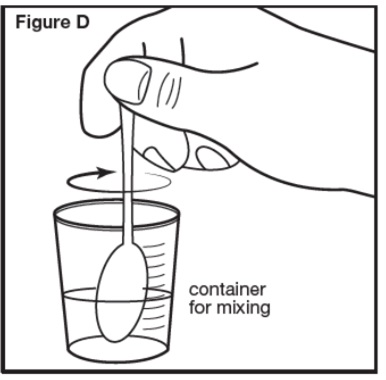
- Stir the contents for 30 seconds or more until the mixture is uniformly mixed.
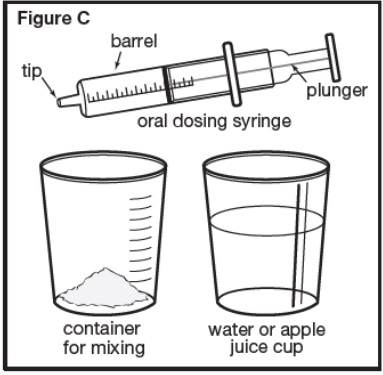
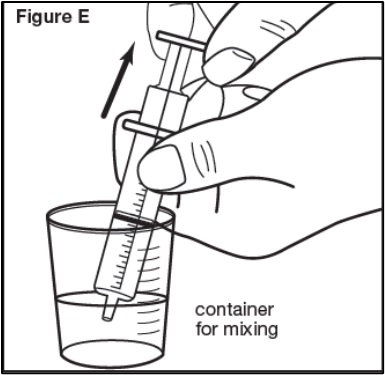
- Using a graduated oral dosing syringe, draw up the prescribed dose volume to the nearest 0.2 mL.
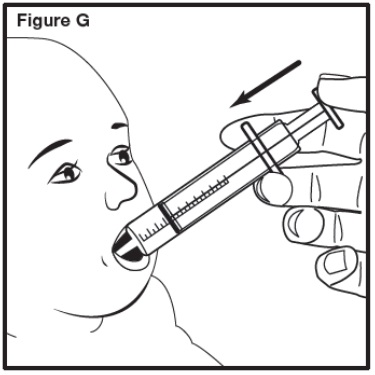
- Administer the prescribed dose volume (mL).
- Administer immediately.
- If particles are remaining in the syringe, draw up additional water or apple juice and administer the contents immediately. Repeat if particles still remain.
- Discard unused portion of SEPHIENCE mixture remaining in the container.
- Consume additional food after administration of the prescribed dose volume.
Doses 1,000 mg or Greater (whole packet administration)
- Determine the required number of SEPHIENCE packets and the required volume of water, apple juice, strawberry jam, or applesauce (see Table 3).
- Prepare a liquid or soft food mixture.
- Open and empty the entire content of each SEPHIENCE packet into a container.
- Mix with the required amount of water, apple juice, strawberry jam, or applesauce per Table 3.
- Stir the contents for 30 seconds or more when mixing SEPHIENCE with water or apple juice or 60 seconds or more when mixing SEPHIENCE with strawberry jam or applesauce until the mixture is uniform.
- Administer the dose.
- Consume the entire mixture immediately.
- If particles are remaining in the container, add additional water or juice to the container and administer the contents immediately. Repeat if particles still remain.
- Consume additional food after administration of the entire mixture.
2.4 Storage Instructions for the Liquid and Soft Food Mixtures
- If the SEPHIENCE liquid or soft food mixture is not administered immediately, cover and store the mixture at controlled room temperature between 20°C to 25°C (68°F to 77°F) for up to 6 hours or refrigerate between 2°C to 8°C (36°F to 46°F) for up to 24 hours.
- If the liquid or soft food mixture is stored, stir for at least 30 or 60 seconds, respectively, prior to administration of the prescribed dose.
- Discard unused SEPHIENCE mixture after 6 hours at controlled room temperature or after 24 hours if refrigerated.
5 WARNINGS AND PRECAUTIONS
- Increased Bleeding: SEPHIENCE may increase the risk of bleeding. Consider treatment interruption in patients with active bleeding. (5.1)
- Hypophenylalaninemia: Some pediatric PKU patients experienced hypophenylalaninemia; monitor patients blood Phe levels during treatment. (5.2)
- Interaction with Levodopa: Seizures, over-stimulation or irritability may occur; monitor patients for a change in neurologic status. (5.3)
5.1 Increased Bleeding
SEPHIENCE may increase the risk of bleeding. Bleeding events, including superficial hematomas, prolonged bleeding, and heavy menstrual bleeding have occurred in patients treated with SEPHIENCE [see Adverse Reactions (6.1)]. One patient with non-traumatic superficial hematomas and prolonged bleeding was re-challenged at a lower dose of SEPHIENCE with recurrence of symptoms, which led to treatment discontinuation. The patient experienced symptoms 15 days after initial exposure and two days after rechallenge. The patient had normal blood counts and coagulation studies at the time of the bleeding. Inform the patient about the increased risk of bleeding associated with SEPHIENCE and to follow up with his/her healthcare provider if he/she experiences any signs of increased bleeding. Consider treatment interruption with SEPHIENCE in patients with active bleeding.
5.2 Hypophenylalaninemia
In clinical trials of SEPHIENCE, some pediatric PKU patients experienced hypophenylalaninemia (low blood Phe), including some patients with multiple low blood Phe levels, during treatment with SEPHIENCE [see Adverse Reactions (6.1)]. Prolonged levels of blood Phe that are too low have been associated with catabolism and endogenous protein breakdown, which has been associated with adverse developmental outcomes.
Monitor blood Phe levels during treatment and if needed, modify the dosage of SEPHIENCE and/or dietary protein and Phe intake to ensure adequate blood Phe level control. Frequent blood Phe monitoring is recommended in the pediatric population [see Dosage and Administration (2.2)].
5.3 Interaction with Levodopa
In a 10-year post-marketing safety surveillance program for a non-PKU indication using another drug that is a phenylalanine hydroxylase (PAH) activator, 3 patients with underlying neurological disorders experienced seizures, exacerbation of seizures, over-stimulation, and irritability during co-administration with levodopa. Monitor patients who are receiving levodopa for changes in neurological status during treatment with SEPHIENCE [see Drug Interactions (7.2)].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
Most common adverse reactions with SEPHIENCE (≥2% and greater than placebo) were diarrhea, headache, abdominal pain, hypophenylalaninemia, feces discoloration, and oropharyngeal pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact PTC Therapeutics, Inc. at 1-866-562-4620 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of SEPHIENCE was evaluated in Trial 1 [Part 1 (open label); Part 2, (placebo-controlled)]; and Trial 2 (open-label). The two trials included a total of 215 SEPHIENCE-treated patients with PKU: 10 (5%) were <2 years old, 118 (55%) were ≥2 and <17 years old, and 87 (40%) were ≥17 years old. All patients received SEPHIENCE from 7.5 mg/kg/day up to 60 mg/kg/day and the median duration of treatment (in weeks) was 25.5 [see Clinical Studies (14.1)].
Trial 1
Trial 1 included a total of 157 patients (85 male and 72 female, aged 1 year to 61 years old) with PKU across both parts of the trial. The patients received dosages from 20 mg/kg up to 60 mg/kg daily and the median duration of treatment was 8 weeks.
Table 4 lists the most common adverse reactions that were reported in ≥2% of patients treated with SEPHIENCE and greater than that of the placebo group in Part 2 of Trial 1.
Adverse reactions were similar across both adult and pediatric populations except for hypophenylalaninemia (see Description of Selected Adverse Reactions ).
Description of Selected Adverse Reactions
Increased Bleeding
In Trial 1 Part 2, a case of heavy menstrual bleeding was reported in a SEPHIENCE-treated patient. Less than 2% of patients in Trial 2 experienced increased bleeding, which included heavy menstrual bleeding, non-traumatic superficial hematomas, and prolonged bleeding.
Hypophenylalaninemia
In Trial 1 Part 2, hypophenylalaninemia was seen in 5% (2/37) of sepiapterin-treated pediatric patients and in no adult patients. Some pediatric patients in Trial 2 had multiple low blood Phe levels.
7 DRUG INTERACTIONS
Dihydrofolate Reductase (DHFR) Inhibitors: Avoid concomitant use (e.g., trimethoprim, methotrexate, trimetrexate, pemetrexed, pralatrexate, raltitrexed, or piritrexim). (7.1)
Sepiapterin Reductase (SR) Inhibitors: Avoid concomitant use (e.g. sulfasalazine or sulfamethoxazole). (7.1)
Interaction with Levodopa: Monitor patients for a change in neurologic status. (7.2)
Drugs Affecting Nitric Oxide-Mediated Vasorelaxation: Potential for vasorelaxation; monitor blood pressure (e.g., PDE-5 inhibitors). (7.2)
7.1 Effects of Other Drugs on SEPHIENCE
Avoid concomitant use of drugs known to inhibit folate synthesis dihydrofolate reductase (DHFR) (e.g., trimethoprim, methotrexate, trimetrexate, pemetrexed, pralatrexate, raltitrexed, and piritrexim) while taking SEPHIENCE. Concomitant administration of such drugs may reduce sepiapterin metabolism to tetrahydrobiopterin (BH4). If concomitant use is not avoidable, monitor blood Phe levels.
Avoid concomitant use of sepiapterin reductase (SR) inhibitors with SEPHIENCE. Concomitant administration of such drugs may reduce sepiapterin metabolism to BH4. If concomitant use is not avoidable, monitor blood Phe levels.
7.2 Effects of SEPHIENCE on Other Drugs
SEPHIENCE may increase the availability of tyrosine, a precursor of levodopa. Neurologic events were reported postmarketing in patients receiving another PAH activator and levodopa concomitantly for a non-PKU indication. Monitor patients for a change in neurologic status when levodopa is administered with SEPHIENCE [see Warnings and Precautions (5.3)].
7.3 Drugs Affecting Nitric Oxide-Mediated Vasorelaxation
SEPHIENCE and PDE-5 inhibitors induce vasorelaxation, thus concomitant use of SEPHIENCE with PDE-5 inhibitors may reduce blood pressure even further. Monitor for signs and symptoms of hypotension with concomitant use of SEPHIENCE with drugs that affect nitric oxide-mediated vasorelaxation (e.g., PDE-5 inhibitors such as sildenafil, vardenafil, or tadalafil).
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data on the use of SEPHIENCE during pregnancy are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Uncontrolled blood Phe concentrations before and during pregnancy are associated with an increased risk of adverse pregnancy outcomes and fetal adverse effects (see Clinical Considerations).
In animal reproduction studies, oral administration of sepiapterin to pregnant rats and rabbits during organogenesis at dose exposures up to 9- and 6- times the human exposure at the maximum recommended human dose (MRHD) of 60 mg/kg, respectively, resulted in no adverse developmental effects (see Data ).
All pregnancies have a background risk of birth defects, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The background risk of major birth defects and miscarriage in pregnant women with PKU who maintain blood Phe concentrations greater than 600 micromol/L during pregnancy is greater than the corresponding background risk for pregnant women without PKU.
Clinical Considerations
Disease-Associated Maternal and/or Embryofetal Risk
Uncontrolled blood Phe concentrations before and during pregnancy are associated with an increased risk of adverse outcomes and fetal adverse effects (see Data ). To reduce the risk of hyperphenylalaninemia-induced fetal adverse effects, blood Phe concentrations should be maintained between 120 and 360 micromol/L during pregnancy and during the 3 months before conception [see Dosage and Administration (2.2)].
Data
Human Data
Available data from the Maternal Phenylketonuria Collaborative Study on 468 pregnancies and 331 live births in pregnant women with PKU demonstrated that uncontrolled Phe concentrations above 600 micromol/L are associated with an increased risk for major birth defects (including microcephaly, major cardiac malformations), intrauterine fetal growth retardation, and future intellectual disability with low IQ.
Animal Data
In an embryo-fetal development study in rats, SEPHIENCE was administered at dose levels of 100, 300, or 1,000 mg/kg/day via oral gavage to pregnant rats during the period of organogenesis from gestation day (GD) 7 to GD 17. There were no maternal or embryo-fetal developmental toxicities noted at doses up to 1,000 mg/kg/day (9-fold the human AUC0-24 at the MRHD).
In an embryo-fetal development study in pregnant rabbits, SEPHIENCE was administered at dose levels of 100, 300, or 1,000 mg/kg/day via oral gavage to pregnant rabbits during the period of organogenesis from gestation day (GD) 7 to GD 19. There were no maternal or embryo-fetal developmental toxicities at doses up to 1,000 mg/kg/day (6-fold the human AUC0-24 at the MRHD).
In the pre- and post-natal development study in rats, SEPHIENCE was administered at dose levels of 30, 100, and 300 mg/kg/day via oral gavage once daily to pregnant rats from GD 6 to lactation day 20. SEPHIENCE did not induce effects on maternal reproductive function or on developmental and reproductive parameters of male and female offspring up to 300 mg/kg (7-fold the human AUC0-24 at the MRHD).
8.2 Lactation
Risk Summary
There are no data on the presence of sepiapterin in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for SEPHIENCE and any potential adverse effects on the breastfed infant from SEPHIENCE or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of SEPHIENCE have been established in pediatric patients 1 month of age and older. Use of SEPHIENCE for this indication is supported by evidence from one adequate and well-controlled trial in 63 pediatric patients with PKU aged 1 to <17 years old (Trial 1) and additional safety and efficacy information obtained from an ongoing open label trial in adult and pediatric patients with PKU (Trial 2) [see Clinical Studies (14.1)].
In Trial 1 Part 2, hypophenylalaninemia was seen in 5.4% (2/37) of sepiapterin-treated pediatric patients and in no adult patients. Some pediatric patients in Trial 2 had multiple low blood Phe levels [see Adverse Reactions (6.1)].
The safety and effectiveness of SEPHIENCE for treatment of PKU have not been established in pediatric patients younger than 1 month of age.
8.5 Geriatric Use
Clinical studies of SEPHIENCE did not include patients 65 years of age and older to determine if they respond differently from younger adult patients.
11 DESCRIPTION
SEPHIENCE (sepiapterin) contains the drug substance sepiapterin, a PAH activator. Sepiapterin is a yellow to orange powder. Sepiapterin is slightly soluble in water with the solubility at 1.4 mg/mL.
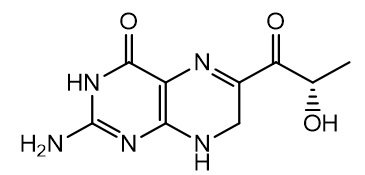
The chemical name of sepiapterin is (S)-2-amino-6-(2-hydroxypropanoyl)-7,8-dihydropteridin-4(3H)-one. The molecular formula is C9H11N5O3 and the molecular weight is 237.22 g/mol. The structural formula is:
SEPHIENCE oral powder contains either 250 mg or 1,000 mg of sepiapterin to be administered orally. The inactive ingredients are: colloidal silicon dioxide, croscarmellose sodium, isomalt, magnesium stearate, mannitol, microcrystalline cellulose, sucralose, and xanthan gum.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
SEPHIENCE is a precursor of the enzymatic co-factor tetrahydrobiopterin (BH4) which activates PAH.
12.2 Pharmacodynamics
Cardiac Electrophysiology
At the recommended SEPHIENCE dose of 60 mg/kg orally once daily, clinically significant QTc interval prolongation was not observed.
12.3 Pharmacokinetics
Following oral administration, sepiapterin reached the maximum concentration in plasma at 2 hours post-treatment and converted to pharmacologically active metabolite BH4 with the exposures of sepiapterin generally less than 2% of those of BH4 (Table 5).
There was no accumulation for BH4 following repeated once daily dose up to 60 mg/kg administered in adult healthy volunteers.
When administered with a high-fat, high-calorie meal in adult healthy volunteers, BH4 exposures increased less than dose proportionally (with slopes 0.87 and 0.84 for Cmax and AUC0-last, respectively) in the dose range 5 to 20 mg/kg and less than dose proportionally (with slopes 0.1957 and 0.3189 for Cmax and AUC0-last, respectively) in the dose range 20 to 60 mg/kg.
The pharmacokinetics of sepiapterin and its active metabolite BH4 following oral administration of sepiapterin at 60 mg/kg with food in adult patients with PKU are summarized in Table 5.
Absorption
The time to maximum plasma concentration (Tmax) of sepiapterin is approximately 1 to 3 hours after oral administration. Plasma sepiapterin is rapidly metabolized to BH4 and peak BH4 concentrations are achieved approximately 4 hours after the oral administration of sepiapterin.
Effect of Food
Administration of SEPHIENCE with food results in increased exposure to sepiapterin and BH4. When SEPHIENCE was administered at 20 or 60 mg/kg daily with a low-fat meal in healthy adult subjects, BH4 exposures were 169% to 172% higher for Cmax and 162% to 173% higher for AUC0-24h compared to administration under fasted conditions. When sepiapterin at 20 or 60 mg/kg was administered with a high-fat, high-calorie meal, BH4 exposures were 221% to 226% higher for Cmax and 251% to 284% higher for AUC0-24h compared to administration under fasted conditions [see Dosage and Administration (2.2)].
Distribution
Sepiapterin mean human plasma protein binding was 15.4% in the presence of 0.1% dithiothreitol (DTT) in the concentration range of 0.1 to 10 μM. BH4 mean human plasma protein binding was between 24.1% and 41.3% in the concentration range 2 to 15 μM in the presence of 0.5% β-mercaptoethanol. Sepiapterin apparent volume of distribution could not be estimated reliably as sepiapterin is converted to BH4 post oral administration and plasma concentration declines to below lower limit of quantitation generally by 12 hours postdose. BH4 apparent volume of distribution is 11433 (5790) L in adult patients with PKU. Increase of BH4 in cerebrospinal fluid was detected after oral administration of sepiapterin 60 mg/kg QD for 7 days in healthy adult subjects.
Elimination
Following oral administration, sepiapterin is quickly absorbed and converted to BH4. Sepiapterin plasma concentration is remarkably lower than BH4 and declines rapidly to below the limit of quantitation generally by 12 hours post dose. The terminal half-life of BH4 is approximately 5 hours and the apparent clearance is 1498 (848) L/h in adult patients with PKU.
Metabolism
Sepiapterin is metabolized by SR/carbonyl reductase (CR) and DHFR in a 2-step unidirectional process to form pharmacologically active metabolite BH4. BH4 is further metabolized non-enzymatically or enzymatically mediated by aromatic amino acid hydroxylases, such as PAH, tyrosine hydroxylase (TH), tryptophan hydroxylase (TPH), pterin-4α-carbinolamine dehydratase (PCD), dihydropteridine reductase (DHPR), xanthine oxidase (XO), and nitric oxide synthase (NOS) in various tissues.
Extensive metabolism of sepiapterin was observed in humans following a single oral dose of 14C-sepiapterin. Absorbed sepiapterin was converted to the active metabolite BH4, with Cmax and AUC0-24h of sepiapterin generally less than 2% of those of BH4.
Excretion
Following a single oral dose of radiolabeled sepiapterin 4,000 mg to healthy adult subjects, a mean of 6.7% was recovered in urine and 26.2% recovered in feces with a combined total recovery of 32.9% by 240 hours. The low total mass recovery is likely due to formation of volatile metabolites in human intestine. Sepiapterin was a minor component in urine and was one of the prominent radioactive components in feces.
Specific Populations
No clinically significant difference in pharmacokinetics of sepiapterin were observed based on age (range 0.5 to 61 years), sex (female 52%, male 48%), race/ethnicity (White 74%, Asian 16%, Other or not specified 7%, or American Indian or Alaska Native 3%) or ABCG2 genotype (BCRP p.Gln141Lys). The effect of renal impairment, hepatic impairment, or pregnancy on pharmacokinetics of sepiapterin or BH4 is unknown.
Pediatric Patients
The pharmacokinetics of sepiapterin and BH4 following oral administration of sepiapterin at 60 mg/kg with food in PKU patients ≥2 years old are summarized in Table 6.
The pharmacokinetics of sepiapterin have not been evaluated in pediatric patients younger than 2 years of age.
Drug Interaction Studies
Both sepiapterin and BH4 were a substrate and inhibitor of efflux transporter breast cancer resistance protein (BCRP) in vitro.
Oral coadministration of curcumin, a BCRP inhibitor, and sepiapterin in healthy adult subjects resulted in increases in mean AUCs and Cmax of BH4 by approximately 20% to 24%, after a single dose.
In healthy adult subjects, oral coadministration of sepiapterin and rosuvastatin, a BCRP substrate, had no impact on rosuvastatin exposures.
The pharmacokinetics of sepiapterin and BH4 following sepiapterin oral administration may be affected by inhibitors of SR and/or DHFR [see Drug Interactions (7.1)].
Sepiapterin may increase the availability of tyrosine, a precursor of levodopa [see Drug Interactions (7.2)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 6-month carcinogenicity study in Tg.rasH2 mice, sepiapterin did not increase the incidence of tumors in male or female transgenic mice at approximately 12- and 16-fold, respectively, the human exposure (AUC0-24h) at the MRHD.
Mutagenesis
Based on the weight of evidence, SEPHIENCE is not genotoxic. SEPHIENCE was negative in the Ames assay. SEPHIENCE was positive in an in vitro chromosomal aberration assay without metabolic activation but not with metabolic activation. SEPHIENCE was negative in the in vivo (micronucleus and comet) assays in rats.
Impairment of Fertility
SEPHIENCE was found to have no effect on fertility and reproductive function of male and female rats when given prior to and throughout mating in male and female rats and continuing to gestation day (GD) 7 in females at oral doses up to 300 mg/kg/day (approximately 7.5-fold the plasma exposure (AUC) at MRHD).
13.2 Animal Toxicology and/or Pharmacology
Repeated administration of SEPHIENCE in rats for 26 weeks at doses greater than 100 mg/kg/day (approximately 3-fold the plasma exposure (AUC) at MRHD) caused renal toxicity, including renal tubular degeneration associated with crystal formation. However, renal toxicity was not noted in monkeys following repeated administration of SEPHIENCE for 39 weeks up to 300 mg/kg/day (approximately 7-fold the plasma exposure (AUC) at MRHD).
14 CLINICAL STUDIES
14.1 Clinical Studies in PKU
Trial 1 (NCT05099640) was a two-part trial in adult and pediatric patients who had a diagnosis of PKU with hyperphenylalaninemia with at least 2 blood Phe measurements ≥600 μmol/L. Patients in the trial were 54% male, 90% White, 5% American Indian or Alaska Native, and 4% Other by race; 16% were identified as Hispanic or Latino. The mean age of the trial patients was 17 years (range: 1 to 61 years). At trial baseline, 8% of patients had blood Phe levels at <360 μmol/L, 36% at 360-600 μmol/L, 48% at 600-1200 μmol/L, and 8% at >1200 μmol/L.
In Part 1, 157 patients received open-label treatment with SEPHIENCE: 7.5 mg/kg orally in patients 0 to <6 months of age, 15 mg/kg in patients 6 to <12 months of age, 30 mg/kg in patients 12 months to <2 years of age, or 60 mg/kg in patients ≥2 years of age per day for 14 days. In Part 1, 66% of PKU patients showed a biochemical response to SEPHIENCE with a >30% or greater reduction in Phe level. In Part 2, after the 2-week washout period from Part 1, 98 patients aged 2 years and older who demonstrated a ≥30% reduction in blood Phe levels to SEPHIENCE treatment in Part 1 were randomized in a double-blind fashion to either SEPHIENCE 20 mg/kg daily for Weeks 1 and 2, 40 mg/kg daily for Weeks 3 and 4, 60 mg/kg daily for Weeks 5 and 6 (N=49), or placebo (N=49) for 6 weeks to assess efficacy. Twelve additional patients who had shown a 15% to 30% reduction in blood Phe during Part 1 were enrolled in Part 2 but were not included in the primary efficacy analysis.
In Part 2, the primary efficacy was assessed in patients who demonstrated a ≥30% reduction in blood Phe levels during Part 1 (N=98) by the mean change in blood Phe level from baseline to Weeks 5 and 6 in the SEPHIENCE-treated group as compared to the mean change in the placebo group, as shown in Table 7.
In Part 2 of Trial 1, 55 patients in the SEPHIENCE group and 54 patients in the placebo group completed the treatment period. One patient in the SEPHIENCE group discontinued treatment.
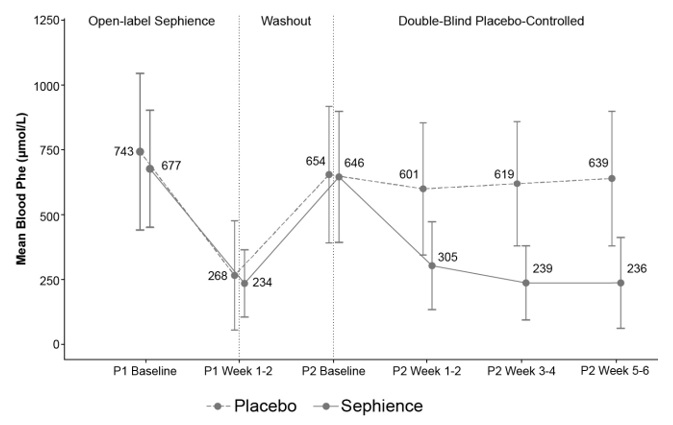
Table 7 displays a reduction in blood Phe level from baseline to Weeks 5 and 6 in Part 2 of Trial 1 for SEPHIENCE-treated patients relative to placebo. Figure 1 displays the mean blood Phe level over time by treatment group.
Figure 1: Mean (±SD) Blood Phe Levels Over Time For Patients Who Had a ≥30% Reduction in Blood Phe Levels During Part 1 of Trial 1 (N=98)
Supportive efficacy data were provided from Trial 2, which is an ongoing, multicenter, open-label trial in adult and pediatric patients with a clinical diagnosis of PKU with hyperphenylalaninemia. At the data cutoff date, 169 patients, including 65 adult and 104 pediatric patients (median age: 14 years, range 2 months to 55 years) received treatment with SEPHIENCE. Five patients 2 to <6 months of age received SEPHIENCE 7.5 mg/kg, 9 patients 12 months to <2 years of age received SEPHIENCE 30 mg/kg, and 90 patients 2 to <17 years of age and 65 patients ≥17 years of age received SEPHIENCE 60 mg/kg. Of the 9 patients under the age of 2 years, 6 patients (66%) had a ≥30% decrease in blood Phe from baseline) at Weeks 1 and 2. The baseline Phe level in patients 2 years and under was 311.4 μmol/L and the mean absolute change in Phe from baseline to Weeks 1 and 2 in this age group was -125 μmol/L (standard deviation 265.9 μmol/L).
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
SEPHIENCE (sepiapterin) oral powder is supplied as a yellow to orange powder in a unit-dose heat-sealed laminated aluminum foil packet. Each packet contains:
250 mg sepiapterin per packet:
NDC 52856-201-03 Carton of 30-unit dose packets
NDC 52856-201-01 Single unit dose packet
1,000 mg sepiapterin per packet:
NDC 52856-301-03 Carton of 30-unit dose packets
NDC 52856-301-01 Single unit dose packet
Storage and Handling
Store SEPHIENCE at room temperature between 20°C to 25°C (68°F to 77°F); excursion permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Store SEPHIENCE liquid or soft food mixture either refrigerated or at controlled room temperature [see Dosage and Administration (2.4)].
17 PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Increased Bleeding
Inform the patient about the increased risk of bleeding associated with SEPHIENCE and to follow up with his/her healthcare provider if he/she experiences any symptoms of increased bleeding.
Drug Interactions
Advise the patient to inform his/her healthcare provider if he/she is taking, or plan to take, any prescription or over-the-counter medications and supplements because of the potential for drug interactions [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
Feces Discoloration
Advise patients that SEPHIENCE may cause feces to have a yellow or orange discoloration.
Manufactured for:
PTC Therapeutics, Inc.
Warren, NJ 07059
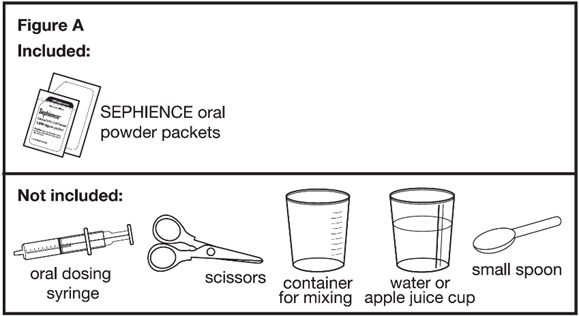
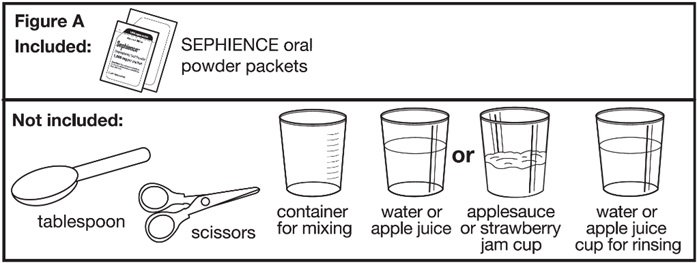


Ask your pharmacist for a container for mixing SEPHIENCE if you do not have these supplies.
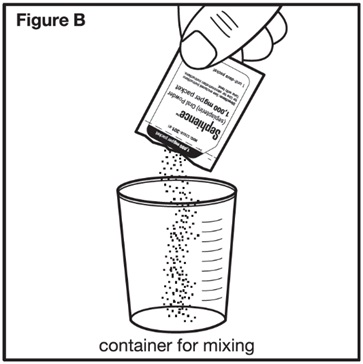
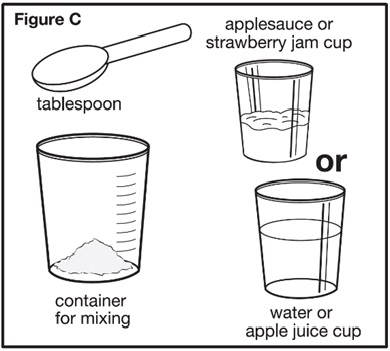
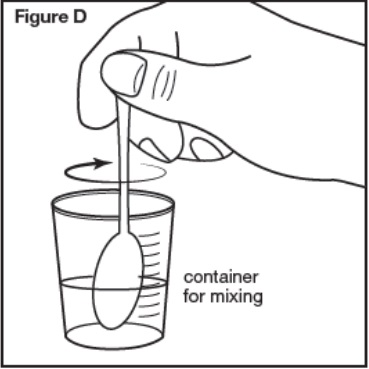
Throwing away (disposing of) SEPHIENCE:
- Ask your pharmacist how to properly throw away (dispose of) SEPHIENCE oral powder packet(s) that are not needed or that have expired.
Manufactured for:
PTC Therapeutics, Inc.
Warren, NJ 07059
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Approved: 4/2026
PRINCIPAL DISPLAY PANEL - NDC: 52856-201-03 - 250mg Carton Label

PRINCIPAL DISPLAY PANEL - NDC: 52856-201-01 - 250mg Sachet Label

PRINCIPAL DISPLAY PANEL - NDC: 52856-201-01 - 250mg Sachet Foil

PRINCIPAL DISPLAY PANEL - NDC: 52856-201-01 - 250mg Sachet Foil

PRINCIPAL DISPLAY PANEL - NDC: 52856-301-03 - 1000mg Carton Label
PRINCIPAL DISPLAY PANEL - NDC: 52856-301-01 - 1000mg Sachet Label
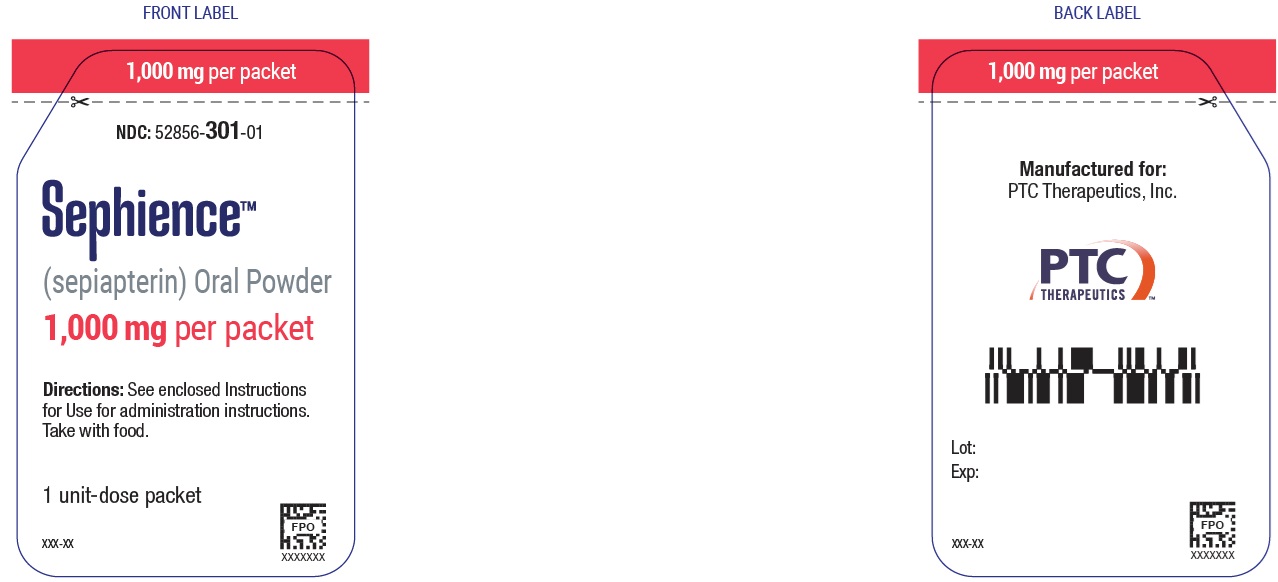
PRINCIPAL DISPLAY PANEL - NDC: 52856-301-01 - 1000mg Sachet Foil

PRINCIPAL DISPLAY PANEL - NDC: 52856-301-01 - 1000mg Sachet Foil

Guideline Central and select third party use “cookies” on this website to enhance the user experience.
This technology helps us gather statistical and analytical information to optimize the relevant content for you.
The user also has the option to opt-out which may have an effect on the browsing experience.