CABOMETYX (cabozantinib) tablet
1 INDICATIONS AND USAGE
CABOMETYX is a kinase inhibitor indicated for the treatment of
- patients with advanced renal cell carcinoma (RCC). (1.1)
- patients with advanced renal cell carcinoma, as a first-line treatment in combination with nivolumab (1.1)
- patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib (1.2)
- adult and pediatric patients 12 years of age and older with locally advanced or metastatic differentiated thyroid cancer (DTC) that has progressed following prior VEGFR-targeted therapy and who are radioactive iodine-refractory or ineligible (1.3)
- adult and pediatric patients 12 years of age and older with previously treated, unresectable, locally advanced or metastatic, well-differentiated pancreatic neuroendocrine tumors (pNET). (1.4)
- adult and pediatric patients 12 years of age and older with previously treated, unresectable, locally advanced or metastatic, well-differentiated extra-pancreatic neuroendocrine tumors (epNET). (1.4)
1.1 Renal Cell Carcinoma
CABOMETYX is indicated for the treatment of patients with advanced renal cell carcinoma (RCC).
CABOMETYX, in combination with nivolumab, is indicated for the first-line treatment of patients with advanced RCC.
1.2 Hepatocellular Carcinoma
CABOMETYX is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib.
1.3 Differentiated Thyroid Cancer
CABOMETYX is indicated for the treatment of adult and pediatric patients 12 years of age and older with locally advanced or metastatic differentiated thyroid cancer (DTC) that has progressed following prior VEGFR-targeted therapy and who are radioactive iodine-refractory or ineligible.
1.4 Neuroendocrine Tumors
CABOMETYX is indicated for the treatment of adult and pediatric patients 12 years of age and older with previously treated, unresectable, locally advanced or metastatic, well-differentiated pancreatic neuroendocrine tumors (pNET).
CABOMETYX is indicated for the treatment of adult and pediatric patients 12 years of age and older with previously treated, unresectable, locally advanced or metastatic, well-differentiated extra-pancreatic neuroendocrine tumors (epNET).
2 DOSAGE AND ADMINISTRATION
- Do NOT substitute CABOMETYX tablets with cabozantinib capsules. (2.1)
- Administer on an empty stomach at least 1 hour before or at least 2 hours after eating. (2.2)
- Stop treatment with CABOMETYX at least 3 weeks prior to scheduled surgery, including dental surgery. (2.1)
- Recommended Dose:
- RCC (Single Agent): 60 mg orally, once daily. (2.2)
- RCC (Combination Therapy): 40 mg orally, once daily with:
- 240 mg nivolumab every 2 weeks by intravenous infusion; -OR-
- 480 mg nivolumab every 4 weeks by intravenous infusion; -OR-
- 600 mg nivolumab and 10,000 units hyaluronidase every 2 weeks administered subcutaneously; -OR-
- 1,200 mg nivolumab and 20,000 units hyaluronidase every 4 weeks administered subcutaneously. (2.2)
- HCC: 60 mg orally, once daily. (2.2)
- DTC, pNET, epNET
- Adult patients and pediatric patients 12 years of age and older with bodyweight greater than or equal to 40 kg: 60 mg orally once daily. (2.2)
- Pediatric patients 12 years of age and older with bodyweight less than 40 kg: 40 mg orally once daily. (2.2)
2.1 Important Dosage Information and Recommended Evaluation and Testing Before Initiating CABOMETYX
- Do not substitute CABOMETYX tablets with cabozantinib capsules.
- Stop treatment with CABOMETYX 3 weeks prior to scheduled surgery, including dental surgery to reduce the risk of hemorrhage. Do not administer CABOMETYX for at least 2 weeks after major surgery and until adequate wound healing [see Warnings and Precautions (5.1, 5.11, 5.12)].
2.2 Recommended Dosage
Administer CABOMETYX on an empty stomach. Administer at least 1 hour before or at least 2 hours after eating [see Clinical Pharmacology (12.3)].
Swallow CABOMETYX tablets whole. Do not crush, chew, or split CABOMETYX tablets.
Do not take a missed dose within 12 hours of the next dose.
Modify the CABOMETYX dose for patients taking drugs known to moderately or strongly induce CYP3A4 or strongly inhibit CYP3A4 and for patients with moderate hepatic impairment [see Dosage and Administration (2.4, 2.5, 2.6)].
The recommended dosages of CABOMETYX are presented in Table 1.
2.3 Dosage Modifications for Adverse Reactions
Withhold CABOMETYX for:
- Intolerable Grade 2 adverse reactions
- Grade 3 or 4 adverse reactions
- Osteonecrosis of the jaw
Upon resolution/improvement (i.e., return to baseline or resolution to Grade 1) of an adverse reaction, reduce the dose as follows:
The following table represents dosage modifications for the drug administered in combination that are different from those described above for CABOMETYX or in the Full Prescribing Information:
When administering CABOMETYX in combination with nivolumab for the treatment of advanced RCC, refer to the nivolumab prescribing information.
2.4 Dosage Modifications for Coadministration with Strong CYP3A4 Inhibitors
Reduce the daily CABOMETYX dose by 20 mg (for example, from 60 mg to 40 mg daily or from 40 mg to 20 mg daily or from 20 mg daily to 20 mg every other day in pediatric patients 12 years of age and older with bodyweight less than 40 kg). Resume the dose that was used prior to initiating the strong CYP3A4 inhibitor 2 to 3 days after discontinuation of the strong inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
2.5 Dosage Modifications for Coadministration with Strong or Moderate CYP3A4 Inducers
Increase the daily CABOMETYX dose by 20 mg (for example, from 60 mg to 80 mg daily or from 40 mg to 60 mg daily) as tolerated. Resume the dose that was used prior to initiating the strong or moderate CYP3A4 inducer 2 to 3 days after discontinuation of the inducer. Do not exceed a daily dose of 80 mg [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
2.6 Dosage Modifications for Patients with Hepatic Impairment
Reduce the starting dose of CABOMETYX 60 mg daily to 40 mg daily or 40 mg daily to 20 mg daily (for pediatric patients 12 years of age and older with bodyweight less than 40 kg) in patients with moderate hepatic impairment (Child-Pugh B) [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
Tablets:
- 60 mg: yellow film-coated, oval shaped with no score, and debossed with "XL" on one side and "60" on the other side.
- 40 mg: yellow film-coated, triangle shaped with no score, and debossed with "XL" on one side and "40" on the other side.
- 20 mg: yellow film-coated, round with no score, and debossed with "XL" on one side and "20" on the other side.
Tablets: 60 mg, 40 mg, 20 mg. (3)
5 WARNINGS AND PRECAUTIONS
- Hemorrhage: Do not administer CABOMETYX if recent history of hemorrhage. (5.1)
- Perforations and Fistulas: Monitor for symptoms. Discontinue CABOMETYX for Grade 4 fistula or perforation. (5.2)
- Thromboembolic Events: Discontinue CABOMETYX for myocardial infarction or serious venous or arterial thromboembolic events. (5.3)
- Hypertension and Hypertensive Crisis: Monitor blood pressure regularly. Interrupt for hypertension that is not adequately controlled with anti-hypertensive therapy. Discontinue CABOMETYX for hypertensive crisis or severe hypertension that cannot be controlled with anti-hypertensive therapy. (5.4)
- Cardiac Failure: Monitor patients for signs and symptoms of cardiac failure throughout treatment. (5.5)
- Diarrhea: May be severe. Interrupt CABOMETYX until diarrhea resolves or decreases to ≤Grade 1, resume at reduced dose. Recommend standard antidiarrheal treatments. (5.6)
- Palmar-Plantar Erythrodysesthesia (PPE): Interrupt CABOMETYX treatment until PPE resolves or decreases to Grade 1. (5.7)
- Hepatotoxicity: When used in combination with nivolumab, higher frequencies of Grade 3 and 4 ALT and AST elevation may occur than with CABOMETYX alone. Monitor liver enzymes before initiation of and periodically throughout treatment. Consider withholding CABOMETYX and/or nivolumab, initiating corticosteroid therapy, and/or permanently discontinuing the combination for severe or life- threatening hepatotoxicity. (5.8)
- Adrenal Insufficiency: When used in combination with nivolumab, primary or secondary adrenal insufficiency may occur. For Grade 2 or higher adrenal insufficiency, initiate symptomatic treatment, including hormone replacement as clinically indicated. Withhold CABOMETYX and/or nivolumab depending on severity. (5.9)
- Proteinuria: Monitor urine protein. Interrupt CABOMETYX until proteinuria resolves to ≤ Grade 1, resume CABOMETYX at a reduced dose. Discontinue for nephrotic syndrome. (5.10)
- Osteonecrosis of the jaw (ONJ): Withhold CABOMETYX for at least 3 weeks prior to invasive dental procedures and for development of ONJ. (5.11)
- Impaired Wound Healing: Withhold CABOMETYX for at least 3 weeks before elective surgery. Do not administer for at least 2 weeks following major surgery and adequate wound healing. The safety of resumption of CABOMETYX after resolution of wound healing complications has not been established. (5.12)
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS): Discontinue CABOMETYX. (5.13)
- Thyroid Dysfunction: Monitor thyroid function before and during treatment with CABOMETYX. (5.14)
- Hypocalcemia: Withhold CABOMETYX and resume at reduced dose upon recovery or permanently discontinue CABOMETYX depending on severity. (5.15)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.16, 8.1, 8.3)
5.1 Hemorrhage
CABOMETYX can cause severe and fatal hemorrhages. Do not administer CABOMETYX to patients who have a recent history of hemorrhage, including hemoptysis, hematemesis, or melena.
The incidence of Grade 3 to 5 hemorrhagic events was 5% in CABOMETYX-treated patients in RCC, HCC, and DTC studies [see Adverse Reactions (6.1)].
Withhold CABOMETYX for 3 weeks prior to scheduled surgery, including dental surgery to reduce the risk of hemorrhage. Permanently discontinue CABOMETYX for Grade 3 or 4 hemorrhage and prior to surgery as recommended [see Dosage and Administration (2.3), Warnings and Precautions (5.11, 5.12)].
5.2 Perforations and Fistulas
CABOMETYX can cause gastrointestinal (GI) perforations and fistulas.
Fistulas, including fatal cases, occurred in 1% of CABOMETYX-treated patients [see [see Adverse Reactions (6.1)]. GI perforations, including fatal cases, occurred in 1% of CABOMETYX-treated patients.
Monitor patients for signs and symptoms of fistulas and perforations, including abscess and sepsis. Discontinue CABOMETYX in patients who experience a Grade 4 fistula or a GI perforation [see Dosage and Administration (2.3)].
5.3 Thromboembolic Events
CABOMETYX can cause arterial or venous thromboembolic events.
Venous thromboembolism occurred in 7% (including 4% pulmonary embolism) and arterial thromboembolism occurred in 2% of CABOMETYX-treated patients. Fatal thromboembolic events occurred in CABOMETYX-treated patients [see Adverse Reactions (6.1)].
Discontinue CABOMETYX in patients who develop an acute myocardial infarction or serious arterial or venous thromboembolic events that require medical intervention [see Dosage and Administration (2.3)].
5.4 Hypertension and Hypertensive Crisis
CABOMETYX can cause hypertension, including hypertensive crisis [see Adverse Reactions (6.1)]. Hypertension was reported in 37% (16% Grade 3 and <1% Grade 4) of CABOMETYX-treated patients. In CABINET (n=195) [see Clinical Studies (14.4)], hypertension was reported in 65% (26% Grade 3) of CABOMETYX-treated patients.
Do not initiate CABOMETYX in patients with uncontrolled hypertension. Monitor blood pressure regularly during CABOMETYX treatment. Withhold CABOMETYX for hypertension that is not adequately controlled with medical management; when controlled, resume CABOMETYX at a reduced dose [see Dosage and Administration (2.3)]. Permanently discontinue CABOMETYX for severe hypertension that cannot be controlled with anti-hypertensive therapy or for hypertensive crisis [see Dosage and Administration (2.3)].
5.5 Cardiac Failure
CABOMETYX can cause severe and fatal cardiac failure [see Adverse Reactions (6.1)]. Cardiac failure occurred in 0.5% of patients treated with CABOMETYX as a single agent, including fatal cardiac failure in 0.1% of patients. Median time to onset of cardiac failure was 73 days (range: 44 days to 159 days).
Consider baseline and periodic evaluations of left ventricular ejection fraction. Monitor for signs and symptoms of cardiovascular events. Withhold and resume at a reduced dose upon recovery or permanently discontinue CABOMETYX depending on the severity [see Dosage and Administration (2.3)].
5.6 Diarrhea
CABOMETYX can cause diarrhea. Diarrhea occurred in 62% of patients treated with CABOMETYX. Grade 3 diarrhea occurred in 10% of patients treated with CABOMETYX [see Adverse Reactions (6.1)].
Monitor and manage patients using antidiarrheals as indicated. Withhold CABOMETYX until improvement to ≤Grade 1, resume CABOMETYX at a reduced dose [see Dosage and Administration (2.3)].
5.7 Palmar-Plantar Erythrodysesthesia
CABOMETYX can cause palmar-plantar erythrodysesthesia (PPE). PPE occurred in 45% of patients treated with CABOMETYX [see Adverse Reactions (6.1)]. Grade 3 PPE occurred in 13% of patients treated with CABOMETYX.
Withhold CABOMETYX until improvement to Grade 1 and resume CABOMETYX at a reduced dose for intolerable Grade 2 PPE or Grade 3 PPE [see Dosage and Administration (2.3)].
5.8 Hepatotoxicity
CABOMETYX in combination with nivolumab can cause hepatic toxicity with higher frequencies of Grades 3 and 4 ALT and AST elevations compared to CABOMETYX alone.
Monitor liver enzymes before initiation of and periodically throughout treatment. Consider more frequent monitoring of liver enzymes as compared to when the drugs are administered as single agents. For elevated liver enzymes, interrupt CABOMETYX and nivolumab and consider administering corticosteroids [see Dosage and Administration (2.3)].
With the combination of CABOMETYX and nivolumab, Grades 3 and 4 increased ALT or AST were seen in 11% of patients [see Adverse Reactions (6.1)]. ALT or AST >3 times ULN (Grade ≥2) was reported in 83 patients, of whom 23 (28%) received systemic corticosteroids; ALT or AST resolved to Grades 0-1 in 74 (89%). Among the 44 patients with Grade ≥2 increased ALT or AST who were rechallenged with either CABOMETYX (n=9) or nivolumab (n=11) as a single agent or with both (n=24), recurrence of Grade ≥2 increased ALT or AST was observed in 2 patients receiving CABOMETYX, 2 patients receiving nivolumab, and 7 patients receiving both CABOMETYX and nivolumab.
Withhold and then resume CABOMETYX at a reduced dose based on severity [see Dosage and Administration (2.3)].
5.9 Adrenal Insufficiency
CABOMETYX in combination with nivolumab can cause primary or secondary adrenal insufficiency.
Adrenal insufficiency occurred in 4.7% (15/320) of patients treated with the combination of CABOMETYX and nivolumab including Grade 3 (2.2%), and Grade 2 (1.9%) adverse reactions [see Adverse Reactions (6.1)]. Adrenal insufficiency led to permanent discontinuation of CABOMETYX and nivolumab in 0.9% and withholding of CABOMETYX and nivolumab in 2.8% of patients with RCC.
Approximately 80% (12/15) of patients with adrenal insufficiency received hormone replacement therapy, including systemic corticosteroids. Adrenal insufficiency resolved in 27% (n=4) of the 15 patients. Of the 9 patients in whom CABOMETYX with nivolumab was withheld for adrenal insufficiency, 6 reinstated treatment after symptom improvement; of these, all (n=6) received hormone replacement therapy and 2 had recurrence of adrenal insufficiency.
For Grade 2 or higher adrenal insufficiency, initiate symptomatic treatment, including hormone replacement as clinically indicated. Withhold CABOMETYX and/or nivolumab and resume CABOMETYX at a reduced dose depending on severity [see Dosage and Administration (2.3)].
5.10 Proteinuria
CABOMETYX can cause proteinuria.
Proteinuria was observed in 8% of patients receiving CABOMETYX [see Adverse Reactions (6.1)].
Monitor urine protein regularly during CABOMETYX treatment. For Grade 2 or 3 proteinuria, withhold CABOMETYX until improvement to ≤Grade 1 proteinuria, resume CABOMETYX at a reduced dose. Discontinue CABOMETYX in patients who develop nephrotic syndrome [see Dosage and Administration (2.3)].
5.11 Osteonecrosis of the Jaw
CABOMETYX can cause osteonecrosis of the jaw (ONJ).
ONJ occurred in <1% of patients treated with CABOMETYX [see Adverse Reactions (6.1)].
ONJ can manifest as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration or erosion, persistent jaw pain or slow healing of the mouth or jaw after dental surgery. Perform an oral examination prior to initiation of CABOMETYX and periodically during CABOMETYX. Advise patients regarding good oral hygiene practices. Withhold CABOMETYX for at least 3 weeks prior to scheduled dental surgery or invasive dental procedures, if possible. Withhold CABOMETYX for development of ONJ until complete resolution, resume at a reduced dose [see Dosage and Administration (2.3)].
5.12 Impaired Wound Healing
CABOMETYX can cause impaired wound healing [see Adverse Reactions (6.1)]. Withhold CABOMETYX for at least 3 weeks prior to elective surgery [see Dosage and Administration (2.1)]. Do not administer CABOMETYX for at least 2 weeks after major surgery and until adequate wound healing. The safety of resumption of CABOMETYX after resolution of wound healing complications has not been established [see Dosage and Administration (2.1, 2.3)].
5.13 Reversible Posterior Leukoencephalopathy Syndrome
CABOMETYX can cause reversible Posterior Leukoencephalopathy Syndrome (RPLS), a syndrome of subcortical vasogenic edema diagnosed by characteristic finding on MRI. Perform an evaluation for RPLS in any patient presenting with seizures, headache, visual disturbances, confusion or altered mental function. Discontinue CABOMETYX in patients who develop RPLS [see Dosage and Administration (2.3)].
5.14 Thyroid Dysfunction
CABOMETYX can cause thyroid dysfunction, primarily hypothyroidism. Thyroid dysfunction occurred in 19% of patients treated with CABOMETYX, including Grade 3 in 0.4% of patients [see Adverse Reactions (6.1)].
Assess patients for signs of thyroid dysfunction prior to the initiation of CABOMETYX and monitor for signs and symptoms of thyroid dysfunction during CABOMETYX treatment. Thyroid function testing and management of dysfunction should be performed as clinically indicated [see Dosage and Administration (2.3)].
5.15 Hypocalcemia
CABOMETYX can cause hypocalcemia. Hypocalcemia occurred in 13% of patients treated with CABOMETYX, including Grade 3 in 2% and Grade 4 in 1% of patients [see Adverse Reactions (6.1)]. Laboratory abnormality data were not collected in CABOSUN and CABINET.
In COSMIC-311 (n=125) [see Clinical Studies (14.3)], hypocalcemia occurred in 36% of patients treated with CABOMETYX, including Grade 3 in 6% and Grade 4 in 3% of patients.
Monitor blood calcium levels and replace calcium as necessary during treatment. Withhold and resume at reduced dose upon recovery or permanently discontinue CABOMETYX depending on severity [see Dosage and Administration (2.3)].
5.16 Embryo-Fetal Toxicity
Based on data from animal studies and its mechanism of action, CABOMETYX can cause fetal harm when administered to a pregnant woman. Cabozantinib administration to pregnant animals during organogenesis resulted in embryolethality at exposures below those occurring clinically at the recommended dose, and in increased incidences of skeletal variations in rats and visceral variations and malformations in rabbits.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with CABOMETYX and for 4 months after the last dose [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed elsewhere in the labeling:
- Hemorrhage [see Warnings and Precautions (5.1)]
- Perforations and Fistulas [see Warnings and Precautions (5.2)]
- Thromboembolic Events [see Warnings and Precautions (5.3)]
- Hypertension and Hypertensive Crisis [see Warnings and Precautions (5.4)]
- Cardiac Failure [see Warnings and Precautions (5.5)]
- Diarrhea [see Warnings and Precautions (5.6)]
- Palmar-plantar Erythrodysesthesia [see Warnings and Precautions (5.7)]
- Hepatotoxicity [see Warnings and Precautions (5.8) ]
- Adrenal Insufficiency [see Warnings and Precautions (5.9) ]
- Proteinuria [see Warnings and Precautions (5.10)]
- Osteonecrosis of the Jaw [see Warnings and Precautions (5.11)]
- Impaired Wound Healing [see Warnings and Precautions (5.12)]
- Reversible Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions (5.13)]
- Thyroid Dysfunction [see Warnings and Precautions (5.14)]
- Hypocalcemia [see Warnings and Precautions (5.15)]
The most common (≥ 20%) adverse reactions are:
- as a single agent: diarrhea, fatigue, PPE, decreased appetite, hypertension, nausea, vomiting, weight decreased, constipation. (6.1)
- in combination with nivolumab: diarrhea, fatigue, hepatotoxicity, PPE, stomatitis, rash, hypertension, hypothyroidism, musculoskeletal pain, decreased appetite, nausea, dysgeusia, abdominal pain, cough, and upper respiratory tract infection. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Exelixis, Inc. at 1-855-500-3935 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS AND PRECAUTIONS section and below reflect exposure to CABOMETYX as a single agent at 60 mg orally once daily until disease progression or unacceptable toxicity in 409 patients with RCC enrolled in a randomized, active-controlled trial (CABOSUN, METEOR), 467 patients with HCC enrolled in a randomized, placebo- controlled trial (CELESTIAL), 125 patients with DTC enrolled in a randomized, placebo- controlled trial (COSMIC-311), 195 patients with pNET or epNET enrolled in a randomized, placebo-controlled trial (CABINET), and at 40 mg CABOMETYX in combination with nivolumab 240 mg/m2 every 2 weeks, in 320 patients with RCC enrolled in a randomized, active- controlled trial (CHECKMATE-9ER).
Renal Cell Carcinoma
METEOR
The safety of CABOMETYX was evaluated in METEOR, a randomized, open-label trial in which 331 patients with advanced renal cell carcinoma received CABOMETYX 60 mg once daily and 322 patients received everolimus 10 mg once daily until disease progression or unacceptable toxicity. Patients on both arms who had disease progression could continue treatment at the discretion of the investigator [see Clinical Studies (14)]. The median duration of treatment was 7.6 months (range 0.3 – 20.5) for patients receiving CABOMETYX and 4.4 months (range 0.21 – 18.9) for patients receiving everolimus.
Adverse reactions which occurred in ≥ 25% of CABOMETYX-treated patients, in order of decreasing frequency, were: diarrhea, fatigue, nausea, decreased appetite, palmar-plantar erythrodysesthesia (PPE), hypertension, vomiting, weight decreased, and constipation. Grade 3-4 adverse reactions and laboratory abnormalities which occurred in ≥ 5% of patients were hypertension, diarrhea, fatigue, PPE, hyponatremia, hypophosphatemia, hypomagnesemia, lymphopenia, anemia, hypokalemia, and increased GGT.
The dose was reduced in 60% of patients receiving CABOMETYX and in 24% of patients receiving everolimus. Twenty percent (20%) of patients received CABOMETYX 20 mg once daily as their lowest dose. The most frequent adverse reactions leading to dose reduction in patients treated with CABOMETYX were: diarrhea, PPE, fatigue, and hypertension. Adverse reactions leading to dose interruption occurred in 70% patients receiving CABOMETYX and in 59% patients receiving everolimus. Adverse reactions led to study treatment discontinuation in 10% of patients receiving CABOMETYX and in 10% of patients receiving everolimus. The most frequent adverse reactions leading to permanent discontinuation in patients treated with CABOMETYX were decreased appetite (2%) and fatigue (1%).
Other clinically relevant adverse reactions (all grades) that were reported in <10% of patients treated with CABOMETYX included: wound complications (2%), cardiac failure (<1%), convulsion (<1%), pancreatitis (<1%), osteonecrosis of the jaw (<1%), and hepatitis cholestatic (<1%).
CABOSUN
The safety of CABOMETYX was evaluated in CABOSUN, a randomized, open-label trial in patients with advanced renal cell carcinoma, in which 78 patients received CABOMETYX 60 mg once daily and 72 patients received sunitinib 50 mg once daily (4 weeks on treatment followed by 2 weeks off), until disease progression or unacceptable toxicity [see Clinical Studies (14.1)]. The median duration of treatment was 6.5 months (range 0.2 – 28.7) for patients receiving CABOMETYX and 3.1 months (range 0.2 – 25.5) for patients receiving sunitinib.
Within 30 days of treatment, there were 4 deaths in patients treated with CABOMETYX and 6 deaths in patients treated with sunitinib. Of the 4 patients treated with CABOMETYX, 2 patients died due to gastrointestinal perforation, 1 patient had acute renal failure, and 1 patient died due to clinical deterioration. All Grade 3-4 adverse reactions were collected in the entire safety population. The most frequent Grade 3-4 adverse reactions (≥5%) in patients treated with CABOMETYX were hypertension, diarrhea, hyponatremia, hypophosphatemia, PPE, fatigue, increased ALT, decreased appetite, stomatitis, pain, hypotension, and syncope.
The median average daily dose was 50.3 mg for CABOMETYX and 44.7 mg for sunitinib (excluding scheduled sunitinib non-dosing days). The dose was reduced in 46% of patients receiving CABOMETYX and in 35% of patients receiving sunitinib. The dose was held in 73% of patients receiving CABOMETYX and in 71% of patients receiving sunitinib. Based on patient disposition, 21% of patients receiving CABOMETYX and 22% of patients receiving sunitinib discontinued due to an adverse reaction.
CHECKMATE-9ER
The safety of CABOMETYX with nivolumab was evaluated in CHECKMATE-9ER, a randomized, open-label study in patients with previously untreated advanced RCC [see Clinical Studies (14.1)]. Patients received CABOMETYX 40 mg orally once daily with nivolumab 240 mg over 30 minutes every 2 weeks (n=320) or sunitinib 50 mg daily, administered orally for 4 weeks on treatment followed by 2 weeks off (n=320) [see Clinical Studies (14.1)]. CABOMETYX could be interrupted or reduced to 20 mg daily or 20 mg every other day. The median duration of treatment was 14 months (range: 0.2 to 27 months) in CABOMETYX and nivolumab-treated patients. In this trial, 82% of patients in the CABOMETYX and nivolumab arm were exposed to treatment for >6 months and 60% of patients were exposed to treatment for >1 year.
Serious adverse reactions occurred in 48% of patients receiving CABOMETYX and nivolumab. The most frequent (≥2%) serious adverse reactions were diarrhea, pneumonia, pneumonitis, pulmonary embolism, urinary tract infection, and hyponatremia. Fatal intestinal perforations occurred in 3 (0.9%) patients.
Adverse reactions leading to discontinuation of either CABOMETYX or nivolumab occurred in 20% of patients: 8% CABOMETYX only, 7% nivolumab only, and 6% both drugs due to the same adverse reaction at the same time. Adverse reactions leading to dose interruption or reduction of either CABOMETYX or nivolumab occurred in 83% of patients: 46% CABOMETYX only, 3% nivolumab only, and 21% both drugs due to the same adverse reaction at the same time, and 6% both drugs sequentially.
The most common adverse reactions reported in ≥20% of patients treated with CABOMETYX and nivolumab were diarrhea, fatigue, hepatotoxicity, PPE, stomatitis, rash, hypertension, hypothyroidism, musculoskeletal pain, decreased appetite, nausea, dysgeusia, abdominal pain, cough, and upper respiratory tract infection.
Hepatocellular Carcinoma
The safety of CABOMETYX was evaluated in CELESTIAL, a randomized, double-blind, placebo-controlled trial in which 704 patients with advanced hepatocellular carcinoma were randomized to receive CABOMETYX 60 mg orally once daily (n=467) or placebo (n=237) until disease progression or unacceptable toxicity [see Clinical Studies (14.2)]. The median duration of treatment was 3.8 months (range 0.1 – 37.3) for patients receiving CABOMETYX and 2.0 months (range 0.0 – 27.2) for patients receiving placebo. The population exposed to CABOMETYX was 81% male, 56% White, and had a median age of 64 years.
Adverse reactions occurring in ≥25% of CABOMETYX-treated patients, in order of decreasing frequency were: diarrhea, decreased appetite, PPE, fatigue, nausea, hypertension, and vomiting. Grade 3-4 adverse reactions which occurred in ≥5% of patients were PPE, hypertension, fatigue, diarrhea, asthenia, and decreased appetite. There were 6 adverse reactions leading to death in patients receiving CABOMETYX (hepatic failure, hepatorenal syndrome, esophagobronchial fistula, portal vein thrombosis, pulmonary embolism, upper gastrointestinal hemorrhage).
The median average daily dose was 35.8 mg for CABOMETYX. The dose was reduced in 62% of patients receiving CABOMETYX; 33% of patients required a reduction to 20 mg daily. The most frequent adverse reactions or laboratory abnormalities leading to dose reduction of CABOMETYX were: PPE, diarrhea, fatigue, hypertension, and increased AST. Adverse reactions leading to dose interruption occurred in 84% patients receiving CABOMETYX. Adverse reactions leading to permanent discontinuation of CABOMETYX occurred in 16% of patients. The most frequent adverse reactions leading to permanent discontinuation of CABOMETYX were PPE (2%), fatigue (2%), decreased appetite (1%), diarrhea (1%), and nausea (1%).
Differentiated Thyroid Cancer
The safety of CABOMETYX was evaluated in COSMIC-311, a randomized, double-blind, placebo-controlled trial in which 187 patients with advanced differentiated thyroid cancer were randomized to receive CABOMETYX 60 mg orally once daily (n=125) or placebo (n=62) with supportive care until disease progression or unacceptable toxicity [see Clinical Studies (14.3)]. At the time of the primary efficacy analysis, the median duration of treatment was 4.4 months (range 0.0 – 15.7) for patients receiving CABOMETYX and 2.3 months (range 0.3 – 11.6) for patients receiving placebo. The median age was 66 years (range 32 to 85 years), 55% were female, 70% were White, 18% were Asian, 2% were Black, 2% were American Indian or Alaska Native, and 63% received prior lenvatinib.
Adverse reactions occurring in ≥25% of CABOMETYX-treated patients, in order of decreasing frequency were: diarrhea, PPE, fatigue, hypertension, and stomatitis. Grade 3-4 adverse reactions which occurred in ≥5% of patients were PPE, hypertension, fatigue, diarrhea, and stomatitis. Serious adverse reactions occurred in 34% of patients who received CABOMETYX. Serious adverse reactions in ≥2% included diarrhea, pleural effusion, pulmonary embolism and dyspnea. Fatal adverse reactions occurred in 1.6% of patients in the CABOMETYX arm, including arterial hemorrhage (0.8%) and pulmonary embolism (0.8%).
The median average daily dose was 42.0 mg for CABOMETYX. The dose was reduced in 56% of patients receiving CABOMETYX; 22% of patients required a second dose reduction. The most frequent adverse reactions (≥5%) leading to dose reduction of CABOMETYX were PPE, diarrhea, fatigue, proteinuria, and decreased appetite. Dose interruptions occurred in 72% patients receiving CABOMETYX. Adverse reactions requiring dosage interruption in ≥5% of patients were PPE, diarrhea, dyspnea, hypertension, decreased appetite and proteinuria. Adverse reactions leading to permanent discontinuation of CABOMETYX occurred in 5% of patients.
Neuroendocrine Tumors
Pancreatic Neuroendocrine Tumors (pNET)
The safety of CABOMETYX was evaluated in adult patients with unresectable, locally advanced or metastatic, well-differentiated neuroendocrine tumors in the CABINET trial [see Clinical Studies (14.4)]. Patients received CABOMETYX 60 mg (n=63) or placebo orally (n=31) once daily until disease progression or unacceptable toxicity. Patients with pNET were required to have disease progression after prior treatment with at least one FDA approved therapy (everolimus, sunitinib or lutetium Lu 177 dotatate), other than somatostatin analogs. The median duration of treatment was 8.3 months (range: 0.1 to 37.8) for patients receiving CABOMETYX and 2.9 months (range: 0.1 to 11.2) for patients receiving placebo.
The median age of patients who received CABOMETYX was 60 years (range: 29 to 79), 57% were male, 86% were White, 6% were Asian, 3.2% were Black, 1.6% were American Indian or Alaska Native, 1.6% were Native Hawaiian or Other Pacific Islanders, and 3.2% were Hispanic or Latino.
Serious adverse reactions occurred in 46% of patients who received CABOMETYX. Serious adverse reactions in ≥2% of patients included thromboembolic events (10%), vomiting (6%), sepsis (4.8%), nausea (4.8%), hypoxia (4.8%), hemorrhage (3.2%), abdominal pain (3.2%), musculoskeletal pain (3.2%), blood bilirubin increased (3.2%), fatigue (3.2%), hyperkalemia (3.2%), and hypertension (3.2%).
Permanent discontinuation of CABOMETYX due to an adverse reaction occurred in 19% of patients. Adverse reactions which resulted in permanent discontinuation of CABOMETYX included thromboembolic events, acute kidney injury, rash, dyspnea, fistulas, hemorrhage, cardiac arrest, musculoskeletal pain, COVID-19 infection, Cushing’s syndrome, pneumonia, proteinuria, and myocardial infarction.
The median average daily dose was 41.4 mg for CABOMETYX. Dosage interruptions of CABOMETYX due to an adverse reaction occurred in 83% of patients. Adverse reactions which required dosage interruption in ≥5% of patients included rash, diarrhea, fatigue, thromboembolic events, nausea, hypertension, increased ALT, blood bilirubin increased, musculoskeletal pain, stomatitis, vomiting, and increased AST.
Dose reductions of CABOMETYX due to an adverse reaction occurred in 49% of patients. Adverse reactions which required dose reductions in ≥5% of patients included rash, fatigue, hypertension, and stomatitis.
The most common adverse reactions occurring in ≥20% of CABOMETYX-treated patients were fatigue, increased AST, increased ALT, hypertension, diarrhea, rash, stomatitis, musculoskeletal pain, hyperglycemia, nausea, platelet count decreased, dysgeusia, neutrophil count decreased, abdominal pain, decreased appetite, hemoglobin decreased, dizziness, hypophosphatemia, hypothyroidism, vomiting, increased ALP, and lymphocyte count decreased.
Table 14 summarizes the adverse reactions in patients with pNET in CABINET.
Clinically relevant adverse reactions in <15% of patients who received CABOMETYX included peripheral neuropathy, hemorrhage, cardiac arrhythmia, hypotension, alopecia, and hair color changes.
Table 15 summarizes the laboratory abnormalities in patients with pNET in CABINET.
Extra-Pancreatic Neuroendocrine Tumors (epNET)
The safety of CABOMETYX was evaluated in adult patients with unresectable, locally advanced or metastatic, well-differentiated neuroendocrine tumors in the CABINET trial [see Clinical Studies (14.4)]. Patients received CABOMETYX 60 mg (n=132) or placebo (n=67) orally once daily until disease progression or unacceptable toxicity. Patients with epNET were required to have disease progression after prior treatment with at least one FDA approved therapy (everolimus or lutetium Lu 177 dotatate), other than somatostatin analogs. The median duration of treatment was 5.4 months (range 0.1 to 32.4) for patients receiving CABOMETYX and 2.8 months (range 0.5 to 22.8) for patients receiving placebo.
The median age was 66 years (range 28 to 86), 55% were female, 86% were White, 7% were Black, 2.3% were Asian, 5% had unknown race or race not reported, and 6% were Hispanic or Latino.
Serious adverse reactions occurred in 44% of patients who received CABOMETYX. Serious adverse reactions in ≥2% included hypertension (6%), abdominal pain (5%), musculoskeletal pain (5%), diarrhea (3.0%), vomiting (3.0%), blood bilirubin increased (3.0%), thromboembolic events (3.0%), nausea (2.3%), hemoglobin decreased (2.3%), muscular weakness (2.3%), fatigue (2.3%), sepsis (2.3%), and syncope (2.3%). Fatal adverse reactions occurred in 4.5% of patients who received CABOMETYX, including hepatic failure, multi-organ dysfunction, gastrointestinal hemorrhage, cardiac arrest, ruptured ascending aortic aneurysm, and sudden death not otherwise specified, occurring in one patient each.
Permanent discontinuation of CABOMETYX due to an adverse reaction occurred in 28% of patients receiving CABOMETYX. Adverse reactions which resulted in permanent discontinuation of CABOMETYX included diarrhea, fatigue, increased AST, increased ALT, blood bilirubin increased, rash, thromboembolic events, hypertension, increased ALP, nausea, and stomatitis.
The median average daily dose was 42.9 mg for CABOMETYX. Dosage interruptions of CABOMETYX due to an adverse reaction occurred in 81% of patients. Adverse reactions which required dosage interruption in ≥5% of patients included diarrhea, fatigue, rash, hypertension, nausea, stomatitis, abdominal pain, increased AST, vomiting, and musculoskeletal pain.
Dose reductions of CABOMETYX due to an adverse reaction occurred in 38% of patients. Adverse reactions which required dose reductions in ≥5% of patients included rash, fatigue, diarrhea, and hypertension.
The most common adverse reactions occurring in ≥20% of CABOMETYX-treated patients were fatigue, increased AST, diarrhea, hypertension, increased ALT, platelet count decreased, rash, stomatitis, nausea, white blood cell count decreased, neutrophil count decreased, musculoskeletal pain, dysgeusia, hypothyroidism, decreased appetite, hemoglobin decreased, hyperglycemia, abdominal pain, increased ALP, lymphocyte count decreased, weight decreased, blood creatinine increased, hypoalbuminemia, blood bilirubin increased, hypocalcemia, hypokalemia, and hypomagnesemia.
Table 16 summarizes the adverse reactions in patients with epNET in CABINET.
Clinically relevant adverse reactions in <15% of patients who received CABOMETYX included cardiac arrhythmia, hemorrhage, thromboembolic events, kidney injury, proteinuria, hypotension, peripheral neuropathy, reversible posterior leukoencephalopathy syndrome, alopecia, hair color changes, and cardiac failure.
Table 17 summarizes the laboratory abnormalities in patients with epNET in CABINET.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of CABOMETYX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Vascular Disorders: Arterial (including aortic) aneurysms, dissections, and rupture
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on CABOMETYX
Strong CYP3A4 Inhibitors
Coadministration of a cabozantinib capsule formulation with a strong CYP3A4 inhibitor increased the exposure of cabozantinib, which may increase the risk of exposure-related adverse reactions [see Clinical Pharmacology (12.3)]. Avoid coadministration of CABOMETYX with strong CYP3A4 inhibitors. Reduce the dosage of CABOMETYX if coadministration with strong CYP3A4 inhibitors cannot be avoided [see Dosage and Administration (2.4)]. Avoid grapefruit or grapefruit juice which may also increase exposure of cabozantinib.
Strong or Moderate CYP3A Inducers
Coadministration of a cabozantinib capsule formulation with a strong CYP3A4 inducer decreased the exposure of cabozantinib, which may reduce efficacy [see Clinical Pharmacology (12.3)]. Avoid coadministration of CABOMETYX with strong or moderate CYP3A4 inducers. Increase the dosage of CABOMETYX if coadministration with strong or moderate CYP3A4 inducers cannot be avoided [see Dosage and Administration (2.5)]. Avoid St. John’s wort which may also decrease exposure of cabozantinib.
8 USE IN SPECIFIC POPULATIONS
- Hepatic Impairment: Reduce the CABOMETYX dosage for patients with moderate hepatic impairment. Avoid in patients with severe hepatic impairment. (2.6, 8.6)
- Lactation: Advise not to breastfeed. (8.2)
- Pediatric Use: Monitor open growth plates in adolescent patients. Consider interrupting or discontinuing CABOMETYX if abnormalities occur. (8.4)
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], CABOMETYX can cause fetal harm when administered to a pregnant woman. There are no available data in pregnant women to inform the drug-associated risk. In animal developmental and reproductive toxicology studies administration of cabozantinib to pregnant rats and rabbits during organogenesis resulted in embryofetal lethality and structural anomalies at exposures that were below those occurring clinically at the recommended dose (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In an embryo-fetal development study in pregnant rats, daily oral administration of cabozantinib throughout organogenesis caused increased embryo-fetal lethality compared to controls at a dose of 0.03 mg/kg (approximately 0.12-fold of human area under the curve [AUC] at the recommended dose). Findings included delayed ossification and skeletal variations at a dose of 0.01 mg/kg/day (approximately 0.04-fold of human AUC at the recommended dose).
In pregnant rabbits, daily oral administration of cabozantinib throughout organogenesis resulted in findings of visceral malformations and variations including reduced spleen size and missing lung lobe at 3 mg/kg (approximately 1.1-fold of the human AUC at the recommended dose).
In a pre- and postnatal study in rats, cabozantinib was administered orally from gestation day 10 through postnatal day 20. Cabozantinib did not produce adverse maternal toxicity or affect pregnancy, parturition or lactation of female rats, and did not affect the survival, growth or postnatal development of the offspring at doses up to 0.3 mg/kg/day (0.05-fold of the maximum recommended clinical dose).
8.2 Lactation
Risk Summary
There is no information regarding the presence of cabozantinib or its metabolites in human milk, or their effects on the breastfed child or milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with CABOMETYX and for 4 months after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating CABOMETYX [see Use in Specific Populations (8.1)].
Contraception
CABOMETYX can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Females
Advise females of reproductive potential to use effective contraception during treatment with CABOMETYX and for 4 months after the final dose.
Infertility
Females and Males
Based on findings in animals, CABOMETYX may impair fertility in females and males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of CABOMETYX for the treatment of differentiated thyroid cancer (DTC) and neuroendocrine tumors (NETs) have been established in pediatric patients aged 12 years and older.
Use of CABOMETYX in pediatric patients aged 12 years and older with DTC and NETs is supported by evidence from adequate and well-controlled studies of CABOMETYX in adults with additional population pharmacokinetic data demonstrating that cabozantinib exposure is within the same range between adults and pediatric patients aged 12 years and older at the recommended dosages [see Dosage and Administration (2.2, 2.3), Adverse Reactions (6.1), Clinical Pharmacology (12.3) and Clinical Studies (14.3, 14.4)].
Physeal widening has been observed in children with open growth plates when treated with CABOMETYX. Based on the limited available data of the effects of CABOMETYX on longitudinal growth, physeal and longitudinal growth monitoring is recommended in children with open growth plates.
The safety and effectiveness of CABOMETYX in pediatric patients less than 12 years of age have not been established.
Juvenile Animal Toxicity Data
Juvenile rats were administered cabozantinib at doses of 1 or 2 mg/kg/day from Postnatal Day 12 (comparable to less than 2 years in humans) through Postnatal Day 35 or 70. Mortalities occurred at doses ≥1 mg/kg/day (approximately 0.16 times the clinical dose of 60 mg/day based on body surface area). Hypoactivity was observed at both doses tested on Postnatal Day 22. Targets were generally similar to those seen in adult animals, occurred at both doses, and included the kidney (nephropathy, glomerulonephritis), reproductive organs, gastrointestinal tract (cystic dilatation and hyperplasia in Brunner’s gland and inflammation of duodenum; and epithelial hyperplasia of colon and cecum), bone marrow (hypocellularity and lymphoid depletion), and liver. Tooth abnormalities and whitening as well as effects on bones including reduced bone mineral content and density, physeal hypertrophy, and decreased cortical bone also occurred at all dose levels. Recovery was not assessed at a dose of 2 mg/kg (approximately 0.32 times the clinical dose of 60 mg based on body surface area) due to high levels of mortality. At the low dose level, effects on bone parameters were partially resolved but effects on the kidney and epididymis/testis persisted after treatment ceased.
8.5 Geriatric Use
In CABOSUN and METEOR, 41% of 409 patients treated with CABOMETYX were age 65 years and older, and 8% were 75 years and older. In CELESTIAL, 49% of 467 patients treated with CABOMETYX were age 65 years and older, and 15% were 75 years and older. In COSMIC-311, 50% of 125 patients treated with CABOMETYX were age 65 years and older, and 12% were 75 years and older. In CABINET, 38% of 63 patients treated with CABOMETYX were age 65 years and older, and 5% were 75 years and older in the pNET cohort, and 55% of 132 patients treated with CABOMETYX were age 65 years and older, and 13% were 75 years and older in the epNET cohort [see Clinical Studies (14)].
No overall differences in safety or effectiveness were observed between these patients and younger patients.
Of the 320 patients with RCC treated with CABOMETYX in combination with nivolumab in CHECKMATE-9ER, 41% were 65 years or older and 9% were 75 years or older [see Clinical Studies (14.1)].
No overall difference in safety was reported between older and younger patients receiving both CABOMETYX and nivolumab.
8.6 Hepatic Impairment
Increased exposure to cabozantinib has been observed in patients with moderate (Child-Pugh B) hepatic impairment. Reduce the CABOMETYX dose in patients with moderate hepatic impairment. Avoid CABOMETYX in patients with severe hepatic impairment (Child-Pugh C), since it has not been studied in this population [see Dosage and Administration (2.2, 2.6), Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dosage adjustment is recommended in patients with mild or moderate renal impairment. There is no experience with CABOMETYX in patients with severe renal impairment [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
One case of overdosage was reported following administration of another formulation of cabozantinib; a patient inadvertently took twice the intended dose for 9 days. The patient suffered Grade 3 memory impairment, Grade 3 mental status changes, Grade 3 cognitive disturbance, Grade 2 weight loss, and Grade 1 increase in BUN. The extent of recovery was not documented.
11 DESCRIPTION
CABOMETYX is the (S)-malate salt of cabozantinib, a kinase inhibitor. Cabozantinib (S)-malate is described chemically as N-(4-(6,7-dimethoxyquinolin-4-yloxy)phenyl)-N'-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide, (2S)-hydroxybutanedioate. The molecular formula is C28H24FN3O5C4H6O5 and the molecular weight is 635.6 Daltons as malate salt. The chemical structure of cabozantinib (S)-malate salt is:

Cabozantinib (S)-malate salt is a white to off-white solid that is practically insoluble in aqueous media.
CABOMETYX (cabozantinib) tablets for oral use are supplied as film-coated tablets containing 20 mg, 40 mg, or 60 mg of cabozantinib, which is equivalent to 25 mg, 51 mg, or 76 mg of cabozantinib (S)-malate, respectively. CABOMETYX also contains the following inactive ingredients: microcrystalline cellulose, lactose anhydrous, hydroxypropyl cellulose, croscarmellose sodium, colloidal silicon dioxide, and magnesium stearate.
The film coating contains hypromellose, titanium dioxide, triacetin, and iron oxide yellow.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
In vitro biochemical and/or cellular assays have shown that cabozantinib inhibits the tyrosine kinase activity of MET, VEGFR-1, -2 and -3, AXL, RET, ROS1, TYRO3, MER, KIT, TRKB, FLT-3, and TIE-2. These receptor tyrosine kinases are involved in both normal cellular function and pathologic processes such as oncogenesis, metastasis, tumor angiogenesis, drug resistance, and maintenance of the tumor microenvironment.
12.2 Pharmacodynamics
The exposure-response or safety relationship for cabozantinib is not fully characterized.
Cardiac Electrophysiology
The effect of cabozantinib on QTc interval was evaluated in a randomized, double-blinded, placebo-controlled trial in patients with medullary thyroid cancer administered a cabozantinib capsule formulation. A mean increase in QTcF of 10 - 15 ms was observed at 4 weeks after initiation. A concentration-QTc relationship could not be definitively established. Changes in cardiac wave form morphology or new rhythms were not observed. No patients in this study had a confirmed QTcF > 500 ms nor did any patients in METEOR, CABOSUN, CELESTIAL, CHECKMATE-9ER, COSMIC-311, or CABINET.
12.3 Pharmacokinetics
Repeat daily dosing of a cabozantinib capsule formulation for 19 days resulted in 4- to 5-fold mean cabozantinib accumulation (based on AUC) compared to a single dose administration; steady state was achieved by Day 15.
Absorption
Median time to peak cabozantinib concentrations (Tmax) ranged from 3 to 4 hours post-dose. A 19% increase in the Cmax of CABOMETYX compared to a cabozantinib capsule formulation was observed following a single 140 mg dose. A less than 10% difference in the AUC was observed between CABOMETYX and a cabozantinib capsule formulation [see Dosage and Administration (2.1)].
Food Effect
Cabozantinib Cmax and AUC increased by 41% and 57%, respectively, following a high-fat meal relative to fasted conditions in healthy subjects administered a single oral dose of a cabozantinib capsule formulation.
Distribution
The oral volume of distribution (Vz/F) of cabozantinib is approximately 319 L. Cabozantinib is highly protein bound in human plasma (≥99.7%).
Elimination
The predicted terminal half-life is approximately 99 hours and the clearance (CL/F) at steady-state is estimated to be 2.2 L/hr.
Metabolism
Cabozantinib is a substrate of CYP3A4 in vitro.
Excretion
Approximately 81% of the total administered radioactivity was recovered within a 48-day collection period following a single dose of radiolabeled 14C-cabozantinib in healthy subjects. Approximately 54% was recovered in feces and 27% in urine. Unchanged cabozantinib accounted for 43% of the total radioactivity in feces and was not detectable in urine following a 72-hour collection.
Specific Populations
The following patient characteristics did not result in a clinically relevant difference in the pharmacokinetics of cabozantinib: age (32-86 years), sex, race (Whites and non-Whites), or mild to moderate renal impairment (eGFR ≥30 mL/min/1.73 m2 as estimated by MDRD (modification of diet in renal disease equation)). The pharmacokinetics of cabozantinib is unknown in patients with eGFR <29 mL/min/1.73m2 as estimated by MDRD equation or requiring dialysis.
Pediatric Patients
The systemic exposures to cabozantinib in pediatric patients 12 years and older at the recommended dosages are expected to be comparable to the exposure in adults at the dose of CABOMETYX 60 mg once daily.
Patients with Hepatic Impairment
Based on a population pharmacokinetic analysis of cabozantinib in healthy subjects and patients with cancer, no clinically significant differences in the mean cabozantinib exposure were observed between subjects with normal liver function (total bilirubin and AST ≤ULN) and those with mild hepatic impairment (total bilirubin ≤ULN and AST >ULN or total bilirubin >1 to 1.5x ULN and any AST value). In a dedicated pharmacokinetic study, cabozantinib exposure (AUC0-INF) increased by 63% in patients with moderate hepatic impairment (Child-Pugh B). Patients with severe hepatic impairment have not been studied [see Dosage and Administration (2.6), Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical Studies
CYP3A4 Inhibitors:
Administration of a strong CYP3A4 inhibitor, ketoconazole (400 mg daily for 27 days), with a cabozantinib capsule formulation to healthy subjects increased single-dose cabozantinib exposure (AUC0-INF) by 38%.
CYP3A4 Inducers:
Administration of a strong CYP3A4 inducer, rifampin (600 mg daily for 31 days), with a cabozantinib capsule formulation to healthy subjects decreased single-dose cabozantinib exposure (AUC0-INF) by 77%.
CYP2C8 Substrates:
No clinically-significant effect on single-dose rosiglitazone (a CYP2C8 substrate) exposure (Cmax and AUC) was observed when co-administered with a cabozantinib capsule formulation at steady-state concentrations.
Gastric Acid Reducing Agents:
No clinically-significant effect on cabozantinib exposure (AUC) was observed following co- administration of the proton pump inhibitor (PPI) esomeprazole (40 mg daily for 6 days) with a single 100 mg dose of a cabozantinib capsule formulation to healthy subjects.
In vitro Studies
CYP Enzymes:
Inhibition of CYP3A4 reduced the formation of the oxidative metabolite by >80%. Inhibition of CYP2C9 had a minimal effect on cabozantinib metabolite formation (i.e., a <20% reduction). Inhibition of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6 and CYP2E1 had no effect on cabozantinib metabolite formation.
Although cabozantinib is an inhibitor of CYP2C8 in vitro, a clinical study of this potential interaction concluded that concurrent use did not result in a clinically relevant effect on CYP2C8 substrate exposure. Given this finding, other less sensitive substrates of pathways affected by cabozantinib in vitro (i.e., CYP2C9, CYP2C19, and CYP3A4) were not evaluated in a clinical study, because, although a clinically relevant exposure effect cannot be ruled out, it is unlikely. Cabozantinib does not inhibit CYP1A2 and CYP2D6 isozymes in vitro.
Cabozantinib is an inducer of CYP1A1 mRNA; however, the clinical relevance of this finding is unknown. Cabozantinib does not induce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 or CYP3A4.
Transporters:
Cabozantinib is an inhibitor, but not a substrate, of P-gp transport activities and has the potential to increase concentrations of co-administered substrates of P-gp. The clinical relevance of this finding is unknown.
Cabozantinib is a substrate of MRP2 in vitro and MRP2 inhibitors have the potential to increase concentrations of cabozantinib. The clinical relevance of this finding is unknown.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of cabozantinib has been evaluated in two species: rasH2 transgenic mice and Sprague-Dawley rats. In the 2-year rat carcinogenicity study, once daily oral administration of cabozantinib resulted in a statistically significant increase in the incidence of malignant/complex malignant pheochromocytoma in combination with benign pheochromocytoma or in benign pheochromocytoma alone in male rats at a dose of 1 mg/kg (approximately 5 times the human exposure by AUC at the recommended 60 mg dose). Cabozantinib was not carcinogenic in a 26-week carcinogenicity study in rasH2 transgenic mice at a slightly higher exposure than the intended human therapeutic exposure.
Cabozantinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay and was not clastogenic in both the in vitro cytogenetic assay using human lymphocytes or in the in vivo mouse micronucleus assay.
Based on nonclinical findings, male and female fertility may be impaired by treatment with CABOMETYX. In a fertility study in which cabozantinib was administered to male and female rats at doses of 1, 2.5, and 5 mg/kg/day, male fertility was significantly compromised at doses equal to or greater than 2.5 mg/kg/day (approximately 13-fold of human AUC at the recommended dose), with a decrease in sperm counts and reproductive organ weights. In females, fertility was significantly reduced at doses equal to or greater than 1 mg/kg/day (5-fold of human AUC at the recommended dose) with a significant decrease in the number of live embryos and a significant increase in pre- and post-implantation losses.
Observations of effects on reproductive tract tissues in general toxicology studies were supportive of effects noted in the dedicated fertility study and included hypospermia and absence of corpora lutea in male and female dogs in a 6-month repeat dose study at plasma exposures (AUC) approximately 0.5-fold (males) and <0.1-fold (females) of those expected in humans at the recommended dose. In addition, female rats administered 5 mg/kg/day for 14 days (approximately 9-fold of human AUC at the recommended dose) exhibited ovarian necrosis.
14 CLINICAL STUDIES
14.1 Renal Cell Carcinoma
Previously Treated with Anti-angiogenic Therapy
The efficacy of CABOMETYX was evaluated in METEOR (NCT01865747), a randomized (1:1), open-label, multicenter trial of CABOMETYX versus everolimus conducted in patients with advanced RCC who had received at least 1 prior anti-angiogenic therapy. Patients had to have a Karnofsky Performance Score (KPS) ≥70%. Patients were stratified by the number of prior VEGFR tyrosine kinase inhibitors (TKIs) and Memorial Sloan Kettering Cancer Center (MSKCC) Risk Group.
Patients were randomized to receive CABOMETYX (N=330) 60 mg orally once daily or everolimus (N=328) 10 mg orally once daily. The majority of the patients were male (75%), with a median age of 62 years. Sixty-nine percent (69%) received only one prior anti-angiogenic therapy. Patient distribution by MSKCC risk groups was 46% favorable (0 risk factors), 42% intermediate (1 risk factor), and 13% poor (2 or 3 risk factors). Fifty-four percent (54%) of patients had 3 or more organs with metastatic disease, including lung (63%), lymph nodes (62%), liver (29%), and bone (22%).
The main efficacy outcome measure was progression-free survival (PFS) assessed by a blinded independent radiology review committee among the first 375 subjects randomized. Other efficacy endpoints were objective response rate (ORR) and overall survival (OS) in the Intent-to- Treat (ITT) population. Tumor assessments were conducted every 8 weeks for the first 12 months, then every 12 weeks thereafter. Patients received treatment until disease progression or experiencing unacceptable toxicity. Patients on both arms who had disease progression could continue treatment at the discretion of the investigator.
Statistically significant improvements in PFS, OS, and ORR were demonstrated for CABOMETYX compared to everolimus. Efficacy results are presented in Tables 18 and 19 and Figures 1 and 2.
Figure 1: Kaplan-Meier Curves of Progression-Free Survival in METEOR (First 375 Randomized)

Figure 2: Kaplan-Meier Curve of Overall Survival in METEOR (ITT)

First-line Treatment
CABOSUN
The efficacy of CABOMETYX was evaluated in CABOSUN (NCT01835158), a randomized (1:1), open-label, multicenter trial of CABOMETYX versus sunitinib conducted in patients with advanced RCC who had not received prior therapy. Patients were randomized to receive CABOMETYX (N=79) 60 mg orally once daily or sunitinib (N=78) 50 mg orally once daily (4 weeks on treatment followed by 2 weeks off) until disease progression or unacceptable toxicity. All patients were required to have intermediate or poor risk disease as defined by the International Metastatic RCC Database Consortium (IMDC) risk group categories. Patients were stratified by IMDC risk group and presence of bone metastases (yes/no).
The majority of patients were male (78%), with a median age of 63 years. Patient distribution by IMDC risk groups was 81% intermediate (1-2 risk factors) and 19% poor (≥3 risk factors). Thirty-six percent (36%) patients had bone metastases. Forty-six percent (46%) of patients were ECOG 0, 41% ECOG 1, and 13% ECOG 2.
The major efficacy outcome measure was progression-free survival (PFS) by a retrospective blinded independent radiology review committee (BIRC).
A statistically significant improvement in PFS, as assessed by a blinded independent radiology review committee, was demonstrated for CABOMETYX compared to sunitinib. Efficacy results are presented in Table 20, Figure 3, and Figure 4.
Figure 3: Kaplan-Meier Curve of Progression-Free Survival in CABOSUN

Figure 4: Kaplan-Meier Curve of Overall Survival in CABOSUN

CHECKMATE-9ER
CHECKMATE-9ER (NCT03141177) was a randomized, open-label study of CABOMETYX combined with nivolumab versus sunitinib in patients with previously untreated advanced RCC. CHECKMATE-9ER excluded patients with autoimmune disease or other medical conditions requiring systemic immunosuppression. Patients were stratified by IMDC prognostic score (favorable vs. intermediate vs. poor), PD-L1 tumor expression (≥1% vs. <1% or indeterminate), and region (US/Canada/Western Europe/Northern Europe vs. Rest of World).
Patients were randomized to CABOMETYX 40mg orally daily and nivolumab 240mg intravenously every 2 weeks (n=323), or sunitinib 50 mg orally daily for the first 4 weeks of a 6-week cycle (4 weeks on treatment followed by 2 weeks off) (n=328). Treatment continued until disease progression per RECIST v1.1 or unacceptable toxicity. Treatment beyond RECIST- defined disease progression was permitted if the patient was clinically stable and considered to be deriving clinical benefit by the investigator. Tumor assessments were performed at baseline, after randomization at Week 12, then every 6 weeks until Week 60, and then every 12 weeks thereafter.
The trial population characteristics were: median age 61 years (range: 28 to 90) with 38% ≥65 years of age and 10% ≥75 years of age. The majority of patients were male (74%) and White (82%) and 23% and 77% of patients had a baseline KPS of 70% to 80% and 90% to 100%, respectively. Patient distribution by IMDC risk categories was 22% favorable, 58% intermediate, and 20% poor.
The major efficacy outcome measure was PFS (BICR assessed). Additional efficacy outcome measures were OS and ORR (BICR assessed). The trial demonstrated a statistically significant improvement in PFS, OS, and ORR for patients randomized to CABOMETYX and nivolumab compared with sunitinib. Consistent results for PFS were observed across pre-specified subgroups of IMDC risk categories and PD-L1 tumor expression status. An updated efficacy analysis was conducted when 271 deaths were observed based on the pre-specified number of deaths for the pre-planned final analysis of OS. Efficacy results are shown in Table 21 and Figures 5 and 6.
Figure 5: Kaplan-Meier Curve of Progression-Free Survival in CHECKMATE-9ER

Figure 6: Kaplan-Meier Curve of Updated Overall Survival in CHECKMATE-9ER

In an exploratory analysis, the updated analysis of OS in patients with IMDC favorable, intermediate, intermediate/poor, and poor risk demonstrated a HR (95% CI) of 1.03 (0.55, 1.92), 0.74 (0.54, 1.01), 0.65 (0.50, 0.85), and 0.49 (0.31, 0.79), respectively.
14.2 Hepatocellular Carcinoma
The efficacy of CABOMETYX was evaluated in CELESTIAL (NCT01908426), a randomized (2:1), double-blind, placebo-controlled, multicenter trial in patients with hepatocellular carcinoma (HCC) who had previously received sorafenib and had Child Pugh Class A liver impairment. Patients were randomized to receive CABOMETYX 60 mg orally once daily or placebo until disease progression or unacceptable toxicity. Randomization was stratified by etiology of disease (hepatitis B virus [HBV] with or without hepatitis C virus [HCV] vs. HCV [without HBV] vs. other [without HBV and HCV]), geographic region (Asia vs. other regions), and presence of extrahepatic spread of disease and/or macrovascular invasion (yes vs. no). The primary efficacy outcome measure was overall survival (OS). Additional outcome measures were progression-free survival (PFS) and objective response rate (ORR), as assessed by investigators per RECIST 1.1. Tumor assessments were conducted every 8 weeks.
In CELESTIAL, a total of 707 patients were randomized, 470 to CABOMETYX and 237 to placebo. The median age was 64 years (range 22 to 86 years), 82% were male, 56% were White and 34% were Asian. Baseline ECOG performance status was 0 (53%) or 1 (47%). The etiology of HCC was attributed to HBV in 38% of patients and HCV in 21%; etiology was attributed to causes other than HBV or HCV in 40%. Macroscopic vascular invasion or extra-hepatic tumor spread was present in 78% of patients and 41% had alpha-fetoprotein (AFP) levels ≥400 mcg/L. All patients received prior sorafenib and 27% received two prior systemic therapy regimens.
Efficacy results are summarized in Table 22, Figure 7, and Figure 8.
Figure 7: Kaplan-Meier Curve of Overall Survival in CELESTIAL

Figure 8: Kaplan-Meier Curve of Progression-Free Survival in CELESTIAL

14.3 Differentiated Thyroid Cancer
The efficacy of CABOMETYX was evaluated in COSMIC-311 (NCT03690388), a randomized (2:1), double-blind, placebo-controlled, multicenter trial in patients with locally advanced or metastatic differentiated thyroid cancer (DTC) that had progressed following prior VEGFR-targeted therapy and were radioactive iodine-refractory or ineligible. Patients were randomized to receive CABOMETYX 60 mg orally once daily or placebo with supportive care until disease progression or unacceptable toxicity. Randomization was stratified by prior receipt of lenvatinib (yes vs. no) and age (≤65 years vs >65 years). Eligible patients randomized to placebo were allowed to cross-over to CABOMETYX upon confirmation of progressive disease by blinded independent radiology review committee (BIRC). The multiple primary efficacy outcome measures were progression-free survival (PFS) in the ITT population, and overall response rate (ORR) in the first 100 randomized patients, as assessed by BIRC per RECIST 1.1. Tumor assessments were conducted every 8 weeks. Overall survival (OS) was a descriptive outcome measure.
The primary analysis of PFS included 187 randomized patients. An updated analysis of PFS was performed and included 258 randomized patients. The median age was 65 years (range 31 to 85 years), 53% were female, 70% were White, 19% were Asian, 2% were Black, 2% were American Indian or Alaska Native, and 63% received prior lenvatinib. Baseline ECOG performance status was 0 (46%) or 1 (54%) and 93% of patients had metastatic disease.
The trial demonstrated a statistically significant improvement in PFS, while it failed to demonstrate a statistically significant improvement in ORR, for patients randomized to CABOMETYX compared with placebo. Efficacy results are summarized in Table 23 and Figure 9.
Figure 9: Kaplan-Meier Curve of Progression-Free Survival in COSMIC-311 (Updated Analysis, N=258)

14.4 Neuroendocrine Tumors
Pancreatic Neuroendocrine Tumors
The efficacy of CABOMETYX for the treatment of pancreatic neuroendocrine tumors (pNET) was evaluated in CABINET (NCT03375320), a randomized, double-blind, placebo-controlled, multicenter study in patients with unresectable, locally advanced or metastatic pNET that had progressed on prior therapy. Eligible patients were required to have been previously treated with at least one FDA approved therapy (everolimus, sunitinib, or lutetium Lu 177 dotatate), other than somatostatin analogs. Patients with active brain metastases or cranial epidural disease, and those who had prior treatment with cabozantinib were excluded. The study also excluded patients with clinically significant gastrointestinal (GI) bleeding, GI abnormalities, and tumor with invasion into the GI tract that may increase the risk for GI bleeding or perforation, and patients with tumor invading or encasing major blood vessels. Patients were randomized (2:1) to receive treatment with CABOMETYX 60 mg orally once daily or placebo until disease progression or unacceptable toxicity. Randomization was stratified by concurrent somatostatin analog (SSA) use (yes/no) and prior sunitinib therapy (yes/no). Tumor assessments were performed every 12 weeks. The study included patients with functional and non-functional tumors, and use of somatostatin analogs at a stable dose was permitted for symptom control. Eligible patients randomized to the placebo arm were allowed to crossover to CABOMETYX upon confirmation of progressive disease by blinded real time central review.
The major efficacy outcome measure was progression-free survival (PFS) assessed by blinded independent radiology review committee (BIRC) per RECIST 1.1. Additional efficacy outcome measures included overall response rate (ORR), duration of response (DOR) and overall survival (OS).
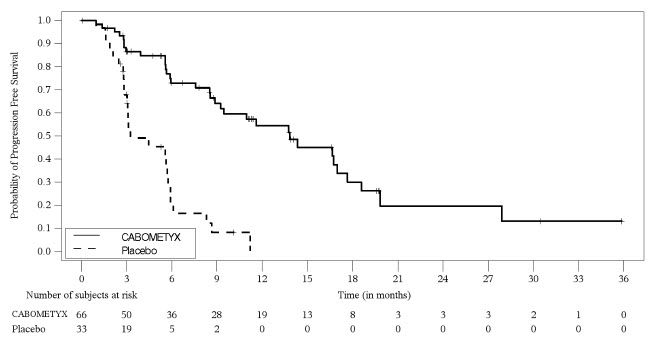
A total of 99 pNET patients were randomized (2:1) to receive CABOMETYX 60 mg orally once daily (n=66) or placebo (n=33). The median age was 60 years (range: 29 to 79); 57% were male; 83% were White, 6% Black or African American, 4.0% Asian, 1.0% American Indian or Alaska Native, 1.0% Native Hawaiian or other Pacific Islander, 1.0% multiple races, 4.0% not reported or unknown; and 5% were Hispanic or Latino. In the 99 patients with pNET, 27% had received one prior systemic therapy, 26% had received two prior systemic therapies and 46% had received three or more prior systemic therapies.
As recommended by the Data and Safety Monitoring Board, the CABINET study was unblinded prior to the final prespecified efficacy analysis and all patients remaining on the placebo arm were permitted to crossover to treatment with cabozantinib. At the time of unblinding, the trial demonstrated a statistically significant improvement in PFS assessed by BIRC for CABOMETYX compared to placebo. An updated OS analysis was conducted when 49 deaths were observed. OS data were not mature with 32 (48%) deaths in CABOMETYX arm and 17 (52%) deaths in placebo arm (OS HR=1.01 [95% CI: 0.55, 1.83]). Fifty-two percent of placebo arm patients crossed over to open label CABOMETYX, which may impact the OS endpoint.
Efficacy results are summarized in Table 24 and Figure 10.
Figure 10: Kaplan-Meier Curve of Progression-Free Survival in Patients with pNET in CABINET
Extra-Pancreatic Neuroendocrine Tumors
The efficacy of CABOMETYX for the treatment of extra-pancreatic neuroendocrine tumors (epNET) was evaluated in CABINET (NCT03375320), a randomized, double-blind, placebo- controlled, multicenter study in patients with unresectable, locally advanced or metastatic epNET that had progressed on prior therapy. Eligible patients were required to have been previously treated with at least one FDA approved therapy (everolimus or lutetium Lu 177 dotatate), other than somatostatin analogs. Patients with active brain metastases or cranial epidural disease, and those who had prior treatment with cabozantinib were excluded. The study also excluded patients with clinically significant gastrointestinal (GI) bleeding, GI abnormalities, and tumor with invasion into the GI tract that may increase the risk for GI bleeding or perforation, and patients with tumor invading or encasing major blood vessels. Patients were randomized (2:1) to receive treatment with CABOMETYX 60 mg orally once daily or placebo until disease progression or unacceptable toxicity. Randomization was stratified by concurrent somatostatin analog (SSA) use (yes/no) and primary site (midgut GI/unknown vs non-midgut GI/lung/other). Tumor assessments were performed every 12 weeks. The study included patients with functional and non-functional tumors, and use of somatostatin analogs at a stable dose was permitted for symptom control. Eligible patients randomized to the placebo arm were allowed to crossover to CABOMETYX upon confirmation of progressive disease by blinded real time central review.
The major efficacy outcome measure was progression-free survival (PFS) assessed by blinded independent radiology review committee (BIRC) per RECIST 1.1. Additional efficacy outcome measures included overall response rate (ORR), duration of response (DOR) and overall survival (OS).
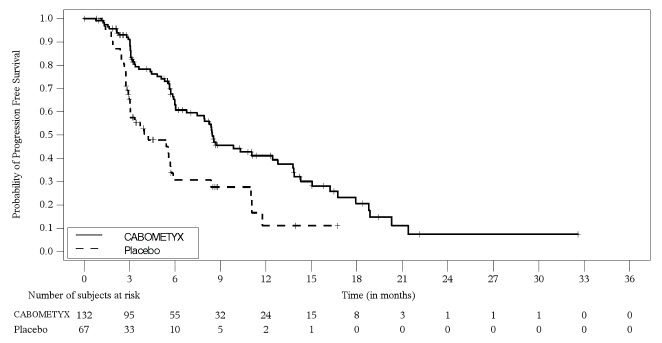
A total of 199 epNET patients were randomized (2:1) to receive CABOMETYX 60 mg orally once daily (n=132) or placebo (n=67). The median age was 66 years (range: 28 to 86), 51% were female; 84% were White; 8% were Black or African American; 2.0% were Asian; 6% had race unknown or race not reported; and 8% were Hispanic or Latino. The primary sites of tumor were small bowel (34%) including duodenum, jejunum & ileum; lung (20%); thymus (5%); rectum (6%); cecum (2.0%); stomach (3.0%); non-cecum colon (1.0%); appendix (0.5%); others (18%); and unknown (12%). In the 199 patients with epNET, 46% had received one prior systemic therapy, 29% had received two prior systemic therapies and 25% had received three or more prior systemic therapies.
The trial demonstrated a statistically significant improvement in PFS as assessed by BIRC, for CABOMETYX compared to placebo. An updated OS analysis was conducted when 123 deaths were observed. OS data were not mature with 83 (63%) deaths in CABOMETYX arm and 40 (60%) deaths in placebo arm (OS HR=1.05 [95% CI: 0.71, 1.54]). Thirty-seven percent of placebo arm patients crossed over to open label CABOMETYX, which may impact the OS endpoint.
Efficacy results are summarized in Table 25 and Figure 11.
Figure 11: Kaplan-Meier Curve of Progression-Free Survival in Patients with epNET in CABINET
16 HOW SUPPLIED/STORAGE AND HANDLING
CABOMETYX tablets are supplied as follows:
60 mg tables are yellow film-coated, oval shaped with no score, debossed with "XL" on one side and "60" on the other side of the tablet; available in:
bottle of 30 tablets: NDC 42388-023-26
bottle of 30 tablets packaged in a carton: NDC 42388-023-46
40 mg tablets are yellow film-coated, triangle shaped with no score, debossed with "XL" on one side and "40" on the other side of the tablet; available in:
bottle of 30 tablets: NDC 42388-025-26
bottle of 30 tablets packaged in a carton: NDC 42388-025-46
20 mg tablets are yellow film-coated, round shaped with no score, debossed with "XL" on one side and "20" on the other side of the tablet; available in:
bottle of 30 tablets: NDC 42388-024-26
bottle of 30 tablets packaged in a carton: NDC 42388-024-46
Store CABOMETYX at 20°C to 25°C (68°F to 77°F); excursions are permitted from 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
- Hemorrhage: Instruct patients to contact their healthcare provider to seek immediate medical attention for signs or symptoms of unusual severe bleeding or hemorrhage [see Warnings and Precautions (5.1)].
- Perforations and fistulas: Advise patients that gastrointestinal disorders such as diarrhea, nausea, vomiting, and constipation may develop during CABOMETYX treatment and to seek immediate medical attention if they experience persistent or severe abdominal pain because cases of gastrointestinal perforation and fistula have been reported in patients taking CABOMETYX [see Warnings and Precautions (5.2)].
- Thromboembolic Events: Venous and arterial thromboembolic events have been reported. Advise patients to report signs or symptoms of an arterial thrombosis. Venous thromboembolic events including pulmonary embolus have been reported. Advise patients to contact their health care provider if new onset of dyspnea, chest pain, or localized limb edema occurs [see Warnings and Precautions (5.3)].
- Hypertension and hypertensive crisis: Inform patients of the signs and symptoms of hypertension. Advise patients to undergo routine blood pressure monitoring and to contact their health care provider if blood pressure is elevated or if they experience signs or symptoms of hypertension [see Warnings and Precautions (5.4)].
- Cardiac Failure: Advise patients that CABOMETYX can cause cardiac failure. Advise patients to immediately contact their healthcare provider for signs or symptoms of cardiac failure [see Warnings and Precautions (5.5)].
- Diarrhea: Advise patients to notify their healthcare provider at the first signs of poorly formed or loose stool or an increased frequency of bowel movements [see Warnings and Precautions (5.6)].
- Palmar-plantar erythrodysesthesia: Advise patients to contact their healthcare provider for progressive or intolerable rash [see Warnings and Precautions (5.7)].
- Hepatotoxicity: Advise patients to contact their healthcare provider immediately for jaundice, severe nausea or vomiting, or easy bruising or bleeding [see Warnings and Precautions (5.8)].
- Adrenal insufficiency: Advise patients receiving with nivolumab to contact their healthcare provider immediately for signs or symptoms of adrenal insufficiency [see Warnings and Precautions (5.9)].
- Proteinuria: Advise patients to contact their healthcare provider for signs or symptoms of proteinuria [see Warnings and Precautions (5.10)].
- Osteonecrosis of the jaw: Advise patients regarding good oral hygiene practices. Advise patients to immediately contact their healthcare provider for signs or symptoms associated with osteonecrosis of the jaw [see Warnings and Precautions (5.11)].
- Impaired wound healing: Advise patients that CABOMETYX may impair wound healing. Advise patients to inform their healthcare provider of any planned surgical procedure [see Dosage and Administration (2.1), Warnings and Precautions (5.12)].
- Reversible posterior leukoencephalopathy syndrome: Advise patients to immediately contact their health care provider for new onset or worsening neurological function [see Warnings and Precautions (5.13)].
- Thyroid dysfunction: Advise patients that CABOMETYX can cause thyroid dysfunction and that their thyroid function should be monitored regularly during treatment. Advise patients to immediately contact their healthcare provider for signs or symptoms of thyroid dysfunction [see Warnings and Precautions (5.14)].
- Hypocalcemia: Advise patients that CABOMETYX can cause low calcium levels and that their serum calcium levels should be monitored regularly during treatment. Advise patients to immediately contact their healthcare provider for signs or symptoms of hypocalcemia [see Warnings and Precautions (5.15)].
-
Embryo-fetal toxicity:
- Advise females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.16), Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception during treatment with CABOMETYX and for 4 months after the final dose [Use in Specific Populations (8.3)].
- Lactation: Advise women not to breastfeed during treatment with CABOMETYX and for 4 months following the last dose [Use in Specific Populations (8.2)].
- Drug interactions: Advise patients to inform their healthcare provider of all prescription or nonprescription medications, vitamins or herbal products. Inform patients to avoid grapefruit, grapefruit juice, and St. John’s wort [see Drug Interactions (7.1)].
Important administration information
- Instruct patients to take CABOMETYX on an empty stomach, at least 1 hour before or at least 2 hours after eating.
Manufactured for Exelixis, Inc. Alameda, CA 94502
Package Label - Bottle - 30 Tablets - 60mg CABOMETYX - Made in Canada
PRINCIPAL DISPLAY PANEL
NDC 42388-023-26
Cabometyx®
(cabozantinib) tablets
60 mg*
Rx Only
30 tablets
Exelixis®

Package Label - Bottle - 30 Tablets - 40mg CABOMETYX - Made in Canada
PRINCIPAL DISPLAY PANEL
NDC 42388-025-26
Cabometyx®
(cabozantinib) tablets
40 mg*
Rx Only
30 tablets
Exelixis®

Package Label - Bottle - 30 Tablets - 20mg CABOMETYX - Made in Canada
PRINCIPAL DISPLAY PANEL
NDC 42388-024-26
Cabometyx®
(cabozantinib) tablets
20 mg*
Rx Only
30 tablets
Exelixis®

Package Label - Professional Sample - Bottle - 15 Tablets - 60mg CABOMETYX
PRINCIPAL DISPLAY PANEL
PROFESSIONAL SAMPLE - NOT FOR SALE
NDC 42388-023-37
Cabometyx®
(cabozantinib) tablets
60 mg*
Rx Only
15 tablets
Exelixis®

Package Label - Professional Sample - Bottle - 15 Tablets - 40mg CABOMETYX
PRINCIPAL DISPLAY PANEL
PROFESSIONAL SAMPLE - NOT FOR SALE
NDC 42388-025-37
Cabometyx®
(cabozantinib) tablets
40 mg*
Rx Only
15 tablets
Exelixis®

Package Label - Professional Sample - Bottle - 15 Tablets - 20mg CABOMETYX
PRINCIPAL DISPLAY PANEL
PROFESSIONAL SAMPLE - NOT FOR SALE
NDC 42388-024-37
Cabometyx®
(cabozantinib) tablets
20 mg*
Rx Only
15 tablets
Exelixis®

Package Label - Bottle - 30 Tablets - 60mg CABOMETYX - Made in Germany
PRINCIPAL DISPLAY PANEL
NDC 42388-023-26
Cabometyx®
(cabozantinib) tablets
60 mg*
Rx Only
30 tablets
Exelixis®

Package Label - Bottle - 30 Tablets - 40mg CABOMETYX - Made in Germany
PRINCIPAL DISPLAY PANEL
NDC 42388-025-26
Cabometyx®
(cabozantinib) tablets
40 mg*
Rx Only
30 tablets
Exelixis®

Package Label - Bottle - 30 Tablets - 20mg CABOMETYX - Made in Germany
PRINCIPAL DISPLAY PANEL
NDC 42388-024-26
Cabometyx®
(cabozantinib) tablets
20 mg*
Rx Only
30 tablets
Exelixis®

Package Carton - 30 Tablets - 60mg CABOMETYX - Made in Canada
PRINCIPAL DISPLAY PANEL
NDC 42388-023-46
Cabometyx®
(cabozantinib) tablets
60 mg*
Rx Only
30 tablets
Exelixis®

Package Carton - 30 Tablets - 40mg CABOMETYX - Made in Canada
PRINCIPAL DISPLAY PANEL
NDC 42388-025-46
Cabometyx®
(cabozantinib) tablets
40 mg*
Rx Only
30 tablets
Exelixis®

Package Carton - 30 Tablets - 20mg CABOMETYX - Made in Canada
PRINCIPAL DISPLAY PANEL
NDC 42388-024-46
Cabometyx®
(cabozantinib) tablets
20 mg*
Rx Only
30 tablets
Exelixis®

Package Carton - Professional Sample - 15 Tablets - 60mg CABOMETYX
PRINCIPAL DISPLAY PANEL
PROFESSIONAL SAMPLE - NOT FOR SALE
NDC 42388-023-57
Cabometyx®
(cabozantinib) tablets
60 mg*
Rx Only
15 tablets
Exelixis®

Package Carton - Professional Sample - 15 Tablets - 40mg CABOMETYX
PRINCIPAL DISPLAY PANEL
PROFESSIONAL SAMPLE - NOT FOR SALE
NDC 42388-025-57
Cabometyx®
(cabozantinib) tablets
40 mg*
Rx Only
15 tablets
Exelixis®

Package Carton - Professional Sample - 15 Tablets - 20mg CABOMETYX
PRINCIPAL DISPLAY PANEL
PROFESSIONAL SAMPLE - NOT FOR SALE
NDC 42388-024-57
Cabometyx®
(cabozantinib) tablets
20 mg*
Rx Only
15 tablets
Exelixis®

Package Carton - 30 Tablets - 60mg CABOMETYX - Made in Germany
PRINCIPAL DISPLAY PANEL
NDC 42388-023-46
Cabometyx®
(cabozantinib) tablets
60 mg*
Rx Only
30 tablets
Exelixis®

Package Carton - 30 Tablets - 40mg CABOMETYX - Made in Germany
PRINCIPAL DISPLAY PANEL
NDC 42388-025-46
Cabometyx®
(cabozantinib) tablets
40 mg*
Rx Only
30 tablets
Exelixis®

Package Carton - 30 Tablets - 20mg CABOMETYX - Made in Germany
PRINCIPAL DISPLAY PANEL
NDC 42388-024-46
Cabometyx®
(cabozantinib) tablets
20 mg*
Rx Only
30 tablets
Exelixis®

Guideline Central and select third party use “cookies” on this website to enhance the user experience.
This technology helps us gather statistical and analytical information to optimize the relevant content for you.
The user also has the option to opt-out which may have an effect on the browsing experience.