DAWNZERA (donidalorsen) injection, solution
1 INDICATIONS AND USAGE
DAWNZERA™ is indicated for prophylaxis to prevent attacks of hereditary angioedema (HAE) in adult and pediatric patients 12 years of age and older.
DAWNZERA is a prekallikrein directed antisense oligonucleotide indicated for prophylaxis to prevent attacks of hereditary angioedema (HAE) in adult and pediatric patients 12 years of age and older. ( 1)
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of DAWNZERA is 80 mg administered subcutaneously every 4 weeks.
- A dosage of 80 mg administered subcutaneously every 8 weeks may be considered.
Missed Dose(s)
If a dose of DAWNZERA is missed, administer DAWNZERA as soon as possible. Resume treatment at the recommended dosing frequency from the date of the most recently administered dose.
2.2 Administration Instructions
- For subcutaneous use.
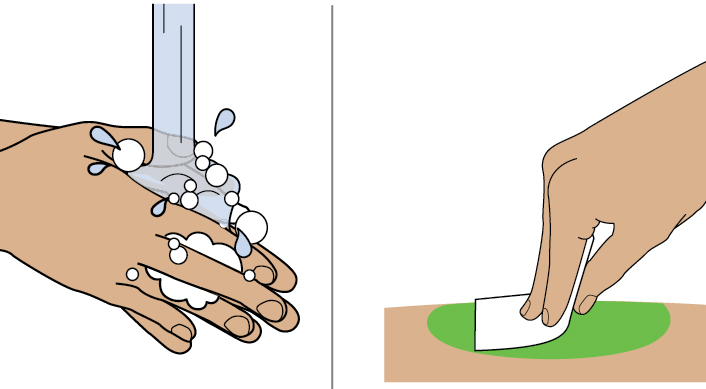
- DAWNZERA is intended for self-administration or administration by a caregiver. Prior to treatment initiation, train patients and/or caregivers on proper preparation and subcutaneous administration technique of DAWNZERA autoinjector [see Instructions for Use] .
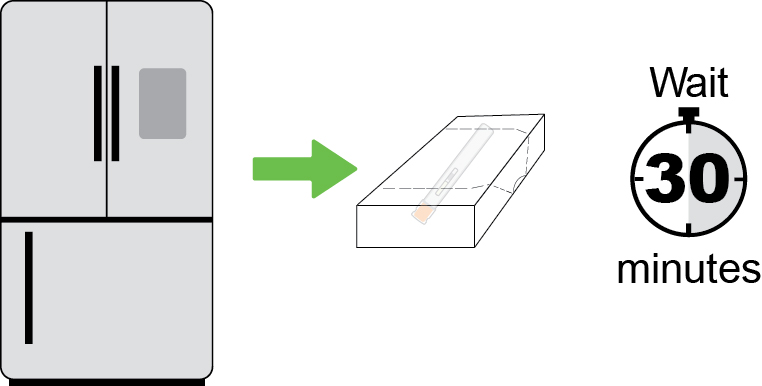
- Remove the single‑dose autoinjector from the refrigerator 30 minutes prior to the injection and allow to warm to room temperature. Do not use other warming methods.
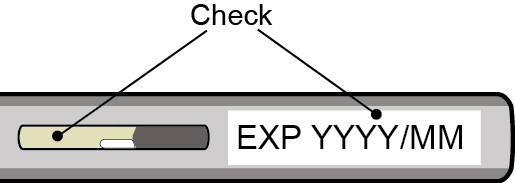
- Inspect DAWNZERA visually for particulate matter and discoloration prior to administration. The solution should appear clear and colorless to yellow. Do not use if cloudiness, particulate matter, or discoloration is observed prior to administration.
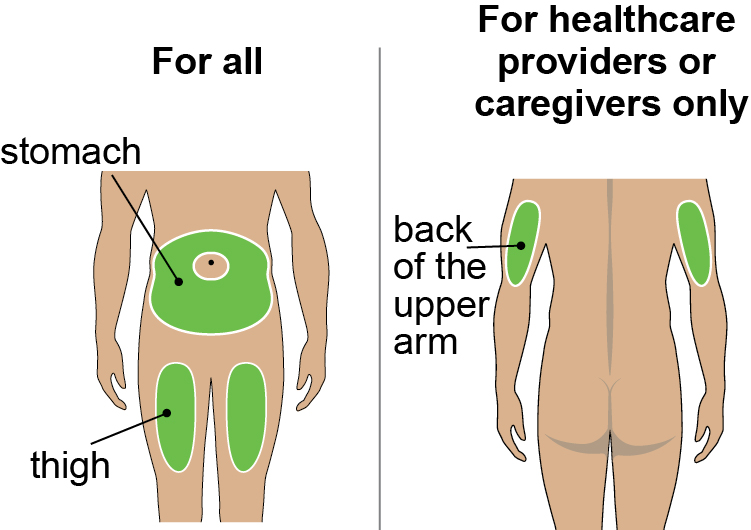
- Administer DAWNZERA subcutaneously into the abdomen or upper thigh region. The back of the upper arm can also be used as an injection site if a caregiver or healthcare provider administers the injection.
3 DOSAGE FORMS AND STRENGTHS
Injection: 80 mg/0.8 mL of donidalorsen as a sterile, clear, colorless to yellow solution in a single-dose autoinjector.
Injection: 80 mg/0.8 mL solution in a single-dose autoinjector. ( 3)
4 CONTRAINDICATIONS
DAWNZERA is contraindicated in patients with a history of serious hypersensitivity reactions, including anaphylaxis, to donidalorsen or any of the excipients in DAWNZERA [see Warnings and Precautions (5.1)and Adverse Reactions (6)] .
History of serious hypersensitivity reactions, including anaphylaxis, to donidalorsen or any of the excipients in DAWNZERA.
5 WARNINGS AND PRECAUTIONS
Hypersensitivity reactions including anaphylaxis have been reported following use of DAWNZERA. Advise patients to discontinue DAWNZERA and seek immediate medical attention if serious hypersensitivity reactions occur. ( 5.1)
5.1 Risk of Hypersensitivity Reactions, Including Anaphylaxis
Hypersensitivity reactions, including anaphylaxis, have been reported in patients treated with DAWNZERA [see Adverse Reactions (6.1)] . If signs and symptoms of serious hypersensitivity reactions occur, discontinue DAWNZERA and institute appropriate therapy.
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed elsewhere in the labeling:
- Risk of Hypersensitivity Reactions, Including Anaphylaxis [see Warnings and Precautions (5.1)]
Most common adverse reactions (incidence ≥ 5%) are injection site reactions, upper respiratory tract infection, urinary tract infection, and abdominal discomfort. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Ionis Pharmaceuticals at 1-833-644-6647 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of DAWNZERA reflects the exposure in a total of 171 adult and pediatric patients 12 years and older with hereditary angioedema (HAE) from a placebo-controlled trial (OASIS-HAE) [see Clinical Studies (14)] , and 2 other clinical studies. The average duration of DAWNZERA treatment exposure across the 3 clinical studies was 14 months.
The safety data below is based on the 24-week multicenter, randomized, double-blind, placebo-controlled trial (OASIS-HAE), in which patients received at least one subcutaneous dose of DAWNZERA 80 mg once every 4 weeks (n=45), DAWNZERA 80 mg once every 8 weeks (n=23), or matching placebo (n=22). Demographics of the patients in OASIS-HAE are summarized in Clinical Studies [see Clinical Studies (14)] .
Table 1 provides the most common adverse reactions with DAWNZERA with incidence ≥5% and more common than placebo.
Specific Adverse Reactions
Hypersensitivity Reactions, Including Anaphylaxis
In clinical trials, hypersensitivity reactions, including anaphylaxis, have occurred. Symptoms included generalized rash, dyspnea, chest pain, and peri-oral swelling.
Laboratory Tests
Decrease in Platelet Count: DAWNZERA can cause reductions in platelet count. In OASIS-HAE, the mean platelet count at baseline was 266,000/mm 3for the DAWNZERA 80 mg every 4 weeks group, 265,000/mm 3for the DAWNZERA 80 mg every 8 weeks group, and 245,000/mm 3for the placebo group. The mean percent change in platelet count at Week 25 was -9.6% for the DAWNZERA 80 mg every 4 weeks group, -7.9% for the DAWNZERA 80 mg every 8 weeks group, and -1.4% for the placebo group. In OASIS-HAE and 2 other clinical studies no DAWNZERA-treated patient had a platelet count of <50,000/mm 3, and there were no major bleeding events associated with a low platelet count.
Increase in Liver Function Tests: Increases from baseline in liver enzymes (alanine aminotransferase, aspartate aminotransferase, and gamma-glutamyl transferase) were observed with DAWNZERA use. The increased levels were generally below 3 times the upper limit of normal and stabilized. Discontinuations due to liver function test increases were infrequent.
8 USE IN SPECIFIC POPULATIONS
Hepatic Impairment: Use in patients with moderate and severe hepatic impairment is not recommended. ( 8.7)
8.1 Pregnancy
Risk Summary
There are no available data on DAWNZERA use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes.
In animal reproduction studies, subcutaneous administration of donidalorsen or a pharmacologically active mouse‑specific surrogate in a combined fertility and embryo‑fetal development study in mice and a pre‑ and postnatal development study in mice with F0 parental doses up to 5 times the maximum recommended human dose (MRHD, 80 mg) on a body surface area (BSA, mg/m 2) basis did not result in any adverse effects on embryofetal development, or behavioral, fertility, and reproductive development in the F1 offspring. Donidalorsen does not cross the placental barrier (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population(s) is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In reproductive toxicity studies with donidalorsen, the unconjugated form was not detected (below the limit of quantitation) in fetal tissues. Donidalorsen does not cross the placental barrier.
In a combined fertility and embryofetal development study, subcutaneous administration of donidalorsen (up to 10 mg/kg/week [2.5-times the MRHD on a BSA basis]) or a mouse‑specific surrogate (4 mg/kg/week) to male and female F0 mice weekly, prior to and during mating, and continuing every other day in females throughout the periods of implantation and organogenesis (Gestation Days 0 to 16), resulted in no adverse effects on embryofetal development. There was no evidence of maternal toxicity with doses up to 10 mg/kg/week.
In a pre- and postnatal development study, subcutaneous administration of donidalorsen (up to 20 mg/kg/week [5-times the MRHD on a BSA basis]) or a mouse-specific surrogate (5 mg/kg/week) to F0 female mice every other day throughout pregnancy (from Gestation Day 6 to 18) and weekly throughout lactation (from Lactation Day 1 to 20) produced no adverse effects on behavioral, fertility, and reproductive development in the F1 offspring. There was no evidence of maternal toxicity with doses up to 20 mg/kg/week.
8.2 Lactation
Risk Summary
There are no data on the presence of donidalorsen in human milk, the effects on the breast‑fed infant, or the effects on milk production. Donidalorsen was excreted into the milk of lactating mice; however, due to species-specific differences in lactation physiology, animal lactation data may not reliably predict levels in human milk (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for DAWNZERA and any potential adverse effects on the breast‑fed infant from DAWNZERA or from the underlying maternal condition.
Data
Animal Data
In the mouse pre‑ and postnatal development study, the concentrations of donidalorsen in breast milk from lactating mice on Lactation Day 15 increased in a dose‑dependent manner at doses ≥10 mg/kg/week, but these concentrations of donidalorsen in breast milk were lower than the observed concentrations in the liver where the drug is preferentially taken up. Even though donidalorsen was detected in the maternal mouse milk, systemic exposure in pups was not expected due to the lack of oral absorption of donidalorsen.
8.4 Pediatric Use
The safety and effectiveness of DAWNZERA for prophylaxis to prevent attacks of HAE have been established in pediatric patients aged 12 years and older. Use of DAWNZERA for this indication is supported by evidence from an adequate and well‑controlled trial (OASIS-HAE) that included 7 pediatric patients (aged 12 to 17 years) who received DAWNZERA 80 mg subcutaneously every 4 weeks (n=4) or every 8 weeks (n=3). The safety and effectiveness of DAWNZERA in pediatric patients aged 12 years and older is extrapolated from adults from OASIS-HAE with support from pharmacokinetic analysis and pharmacodynamic response [see Clinical Pharmacology (12.2, 12.3)and Clinical Studies (14)] . No new safety signals were identified in pediatric patients aged 12 years and older who received DAWNZERA [see Adverse Reactions (6.1)] .
The safety and effectiveness of DAWNZERA have not been established in pediatric patients younger than 12 years of age.
8.5 Geriatric Use
Clinical studies of DAWNZERA did not include sufficient numbers of patients 65 years of age and older to determine whether they respond differently from younger adult patients.
8.6 Renal Impairment
No dosage adjustment of DAWNZERA is recommended for patients with mild renal impairment (estimated glomerular filtration rate [eGFR] ≥60 to <90 mL/min/1.73 m 2) [see Clinical Pharmacology (12.3)] .
DAWNZERA has not been studied in patients with moderate or severe renal impairment or end‑stage renal disease.
8.7 Hepatic Impairment
No dosage adjustment of DAWNZERA is required for patients with mild hepatic impairment (defined by National Cancer Institute Organ Dysfunction Working Group [NCI-ODWG] Criteria: total bilirubin ≤1 × upper limit of normal [ULN] and aspartate aminotransferase [AST] >1 × ULN, or total bilirubin >1 to 1.5 × ULN and any AST level) [see Clinical Pharmacology (12.3)] .
DAWNZERA has not been studied in patients with moderate or severe hepatic impairment. Use of DAWNZERA is not recommended in patients with moderate or severe hepatic impairment (defined by NCI-ODWG Criteria: total bilirubin >1.5 x ULN regardless of AST level).
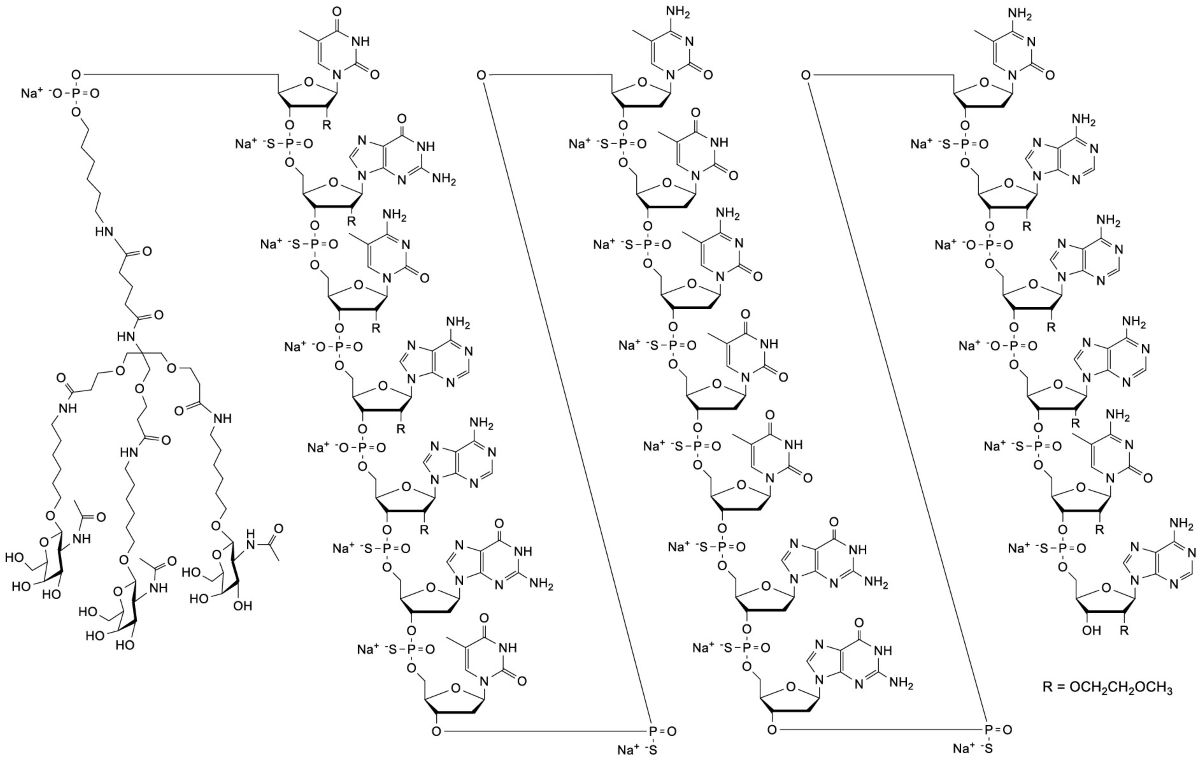
11 DESCRIPTION
Donidalorsen is a prekallikrein-directed antisense oligonucleotide (ASO) covalently linked to a ligand containing three N‑acetyl galactosamine (GalNAc) residues to facilitate delivery of the ASO to hepatocytes.
DAWNZERA contains donidalorsen sodium as the active ingredient. Donidalorsen sodium is a white to yellow solid and it is freely soluble in water and in sodium phosphate buffer. The molecular formula of donidalorsen sodium is C 296H 415N 83O 151P 20S 15Na 20and the molecular weight is 9112.27 daltons. The chemical name of donidalorsen is DNA, d([2′- O-(2-methoxyethyl)]m 5rU-s p-[2′- O-(2-methoxyethyl)]rG-s p-[2′- O-(2-methoxyethyl)]m 5rC-[2′- O-(2-methoxyethyl)]rA-[2′- O-(2-methoxyethyl)]rA-s p-G-s p-T-s p-m 5C-s p-T-s p-m 5C-s p-T-s p-T-s p-G-s p-G-s p-m 5C-s p-[2′- O-(2-methoxyethyl)]rA-[2′- O-(2-methoxyethyl)]rA-[2′- O-(2methoxyethyl)]rA-s p-[2′- O-(2-methoxyethyl)]m 5rC-s p-[2′- O-(2-methoxyethyl)]rA), 5′-[26-[[2-(acetylamino)-2-deoxy-β-d-galactopyranosyl]oxy]-14,14-bis[[3-[[6-[[2-(acetylamino)-2-deoxy-β-d-galactopyranosyl]oxy]hexyl]amino]-3-oxopropoxy]methyl]-8,12,19-trioxo-16-oxa-7,13,20-triazahexacos-1-yl hydrogen phosphate], sodium salt (1:20).
The chemical structure of donidalorsen sodium is presented below:
DAWNZERA (donidalorsen) injection is a sterile, preservative‑free solution for subcutaneous injection supplied as a single-dose autoinjector. Each single‑dose autoinjector contains 80 mg of donidalorsen (equivalent to 84 mg donidalorsen sodium) in 0.8 mL of solution. The solution also contains disodium hydrogen phosphate; sodium chloride; sodium dihydrogen phosphate; water for injection; and may include hydrochloric acid and/or sodium hydroxide for pH adjustment between 6.9 to 7.9. Each dose of DAWNZERA injection contains 6 mg of phosphorous and 5 mg of sodium.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Donidalorsen is an ASO‑GalNAc conjugate that causes ribonuclease H1 (RNase H1)‑mediated degradation of PKK mRNA through binding to PKK mRNA, which results in reduced production of PKK protein. PKK is a pro‑enzyme for plasma kallikrein, which results in the release of bradykinin, a potent vasodilator causing swelling and pain in HAE. In patients with HAE, C1‑inhibitor (C1‑INH) deficiency or dysfunction leads to excessive plasma kallikrein activity, bradykinin generation, and angioedema attacks. Donidalorsen lowers PKK concentration, preventing excessive bradykinin production in patients with HAE.
12.2 Pharmacodynamics
In OASIS-HAE in adult and pediatric patients (≥12 years) with HAE‑1 or HAE‑2 [see Clinical Studies (14)] , a decrease in plasma PKK concentrations was observed at the first assessment (Week 4) following treatment with DAWNZERA 80 mg. The mean percentage reduction from baseline at Week 4 across both treatment groups was 48%. The mean percentage change from baseline to Week 24 in trough plasma PKK concentrations indicated reductions of 73% and 47% following treatment with DAWNZERA 80 mg every 4 weeks and every 8 weeks, respectively, compared with a slight increase (2%) observed in the placebo group.
Cardiac Electrophysiology
At the maximum recommended dose of DAWNZERA 80 mg every 4 weeks, clinically significant QTc interval prolongation was not observed.
12.3 Pharmacokinetics
The pharmacokinetic properties of DAWNZERA were evaluated following subcutaneous administration of multiple doses every 4 weeks in healthy subjects and every 4 weeks or every 8 weeks in patients with HAE. The pharmacokinetics of DAWNZERA were similar between healthy subjects and patients with HAE.
Donidalorsen exposure (area under the plasma concentration‑time curve [AUC]) at steady state following subcutaneous administration in healthy subjects increased in a greater than dose‑proportional manner over the dose range of 0.25 times the maximum recommended dosage to 80 mg every 4 weeks.
Geometric Mean (Coefficient of Variation [CV%]) of steady‑state maximum plasma concentration (C max,ss), trough plasma concentration (C trough,ss), and area under the plasma concentration‑time curve over the dosing interval (AUC τ ,ss) are presented in Table 2. No accumulation of donidalorsen C maxand AUC was observed in plasma after repeated dosing every 4 weeks. However, a 2-fold increase of plasma donidalorsen C troughwas observed following repeated dosing every 4 weeks.
Absorption
Following subcutaneous administration, donidalorsen is absorbed with the median (range) time to maximum plasma concentration of approximately 2 (0.25, 8) hours post dose.
Distribution
Donidalorsen is expected to distribute primarily to the liver and kidney cortex after subcutaneous dosing. The apparent volume of distribution for the central (V c/F) and peripheral (V p/F) compartment were 69.8 L and 1840 L, respectively. Donidalorsen is highly bound to human plasma proteins (>98% bound) in vitro.
Elimination
The terminal plasma elimination half-life of donidalorsen in a typical patient with HAE is approximately 1 month. The half-life of the initial rapid clearance phase, reflecting tissue distribution, was approximately 5 hours.
Metabolism
The oligonucleotide moiety of donidalorsen is expected to be metabolized by endo‑ and exonucleases to short oligonucleotide fragments of varying sizes within the liver. Based on in vitrostudies, donidalorsen is not a substrate of cytochrome P450 (CYP) enzymes.
The linker that covalently connects the ASO to the GalNAc residues is cleaved via hydrolysis and undergoes dephosphorylation and subsequent oxidative metabolism to form inactive metabolites, which are minimally released in circulation. The most abundant linker-related metabolite (M8) is a substrate of CYP3A4.
Excretion
The mean fraction of unchanged ASO eliminated in urine was less than 1% of the administered dose in healthy subjects within 24 hours post-dose. The renal route of elimination is minor for linker-related metabolites.
Specific Populations
No clinically meaningful differences in the pharmacokinetics or pharmacodynamics of donidalorsen were observed based on age (12 to 68 years), body weight (37 to 152 kg), sex, race (68% White, 24% Black, and 4% Asian), ethnicity, disease status (healthy subjects or subjects with HAE), mild renal impairment (eGFR ≥60 to <90 mL/min/1.73 m 2), or mild hepatic impairment (defined using NCI-ODWG Criteria: total bilirubin ≤1 × ULN and AST >1 × ULN, or total bilirubin >1 to 1.5 × ULN and any AST).
Donidalorsen has not been studied in patients with moderate or severe renal impairment, end‑stage renal disease, or moderate or severe hepatic impairment.
Drug Interaction Studies
No clinical drug‑drug interaction studies have been performed with donidalorsen. In vitrostudies show that donidalorsen is not a substrate or inhibitor of transporters, does not interact with highly plasma protein bound drugs, and is not an inhibitor/inducer of CYP enzymes. In vitro studies show that linker-related metabolite M8 is not an inhibitor or inducer of CYP enzymes. M8 is a substrate of transporters bile salt export pump (BSEP) and organic anion transporting polypeptide 1B3 (OATP1B3), and is an inhibitor of multidrug and toxin extrusion protein 1 (MATE1) transporter.
12.6 Immunogenicity
The observed incidence of anti‑drug antibodies (ADAs) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of donidalorsen or of other donidalorsen products.
In OASIS-HAE, with a treatment duration up to 24 weeks, the incidence rate of treatment‑emergent ADAs in adult and pediatric patients (≥12 years of age) with HAE was 20% (9 of 45 patients) in the DAWNZERA 80 mg every 4 weeks group and 22% (5 of 23 patients) in the DAWNZERA 80 mg every 8 weeks group. In an open‑label extension trial, patients that rolled over from OASIS-HAE continued treatment with DAWNZERA in the 80 mg every 4 weeks or every 8 weeks groups for up to 3 years (median exposure duration of 227 days). The incidence rate of treatment‑emergent ADAs was 35% (22 of 63 patients) in the DAWNZERA 80 mg every 4 weeks group, including patients initially randomized to DAWNZERA 80 mg every 4 weeks in OASIS-HAE (36%, 16/44) and patients initially randomized to placebo in OASIS-HAE (32%, 6/19). The incidence rate of treatment-emergent ADAs was 21% (3 of 14 patients) among patients who received DAWNZERA 80 mg every 8 weeks in OASIS-HAE and open-label extension.
In general, the development of ADAs was not found to affect the pharmacodynamics, safety, or efficacy of DAWNZERA. An increase in donidalorsen plasma C troughwas observed in ADA-positive patients with high titers. Because of small sample size, the effect of ADA on the pharmacokinetics, pharmacodynamics, safety and effectiveness of DAWNZERA is inconclusive.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 6-month carcinogenicity study in transgenic (Tg.rasH2) mice, subcutaneous administration of donidalorsen, up to the highest dose tested (20 mg/kg in males and 60 mg/kg in females) or a mouse-specific surrogate (10 mg/kg) once every 2 weeks did not result in an increase in malignant tumors.
Mutagenesis
Donidalorsen was negative for genotoxicity in the in vitrobacterial reverse mutation test and chromosomal aberration assay in Chinese hamster lung cells and in vivomouse bone marrow micronucleus assay.
Fertility
Fertility and reproductive performance were unaffected by subcutaneous administration of donidalorsen (up to 10 mg/kg/week [2.5-times the MRHD on a BSA basis]) or a mouse‑specific surrogate (4 mg/kg/week) to male and female mice weekly, prior to and during mating, and continuing every other day in females throughout the periods of implantation and organogenesis.
14 CLINICAL STUDIES
The efficacy of DAWNZERA for prophylaxis to prevent attacks of hereditary angioedema (HAE) in adult and pediatric patients 12 years of age and older was evaluated in a 24‑week multicenter, randomized, double‑blind, placebo‑controlled trial (OASIS-HAE [ NCT05139810]).
The trial (OASIS-HAE) included 90 adult and pediatric patients 12 years of age and older with Type I and Type II HAE, who had at least 2 investigator‑confirmed attacks during the 8‑week run‑in period. Patients were randomized to receive DAWNZERA 80 mg once every 4 weeks (n=45), DAWNZERA 80 mg once every 8 weeks (n=23), or matching placebo (n=22). Patients were required to discontinue other prophylactic HAE medications, except androgens and tranexamic acid, prior to entering the trial; all patients were allowed to use rescue medications for treatment of breakthrough HAE attacks.
The demographics and baseline characteristics of OASIS-HAE trial are provided in Table 3.
The primary endpoint for OASIS-HAE was the HAE attack rate (number of investigator-confirmed HAE attacks per 4 weeks) from Week 0 to Week 24. As shown in Table 4, DAWNZERA 80 mg administered subcutaneously every 4 or 8 weeks demonstrated statistically significant reductions in the HAE attack rate compared to placebo.
The mean decreases from baseline in the HAE attack rate observed throughout the treatment period in the DAWNZERA treatment groups are shown in Figure 1.
Figure 1: Mean (± SEM) Investigator‑confirmed HAE Attack Rate (Attacks/4 Weeks) in OASIS-HAE
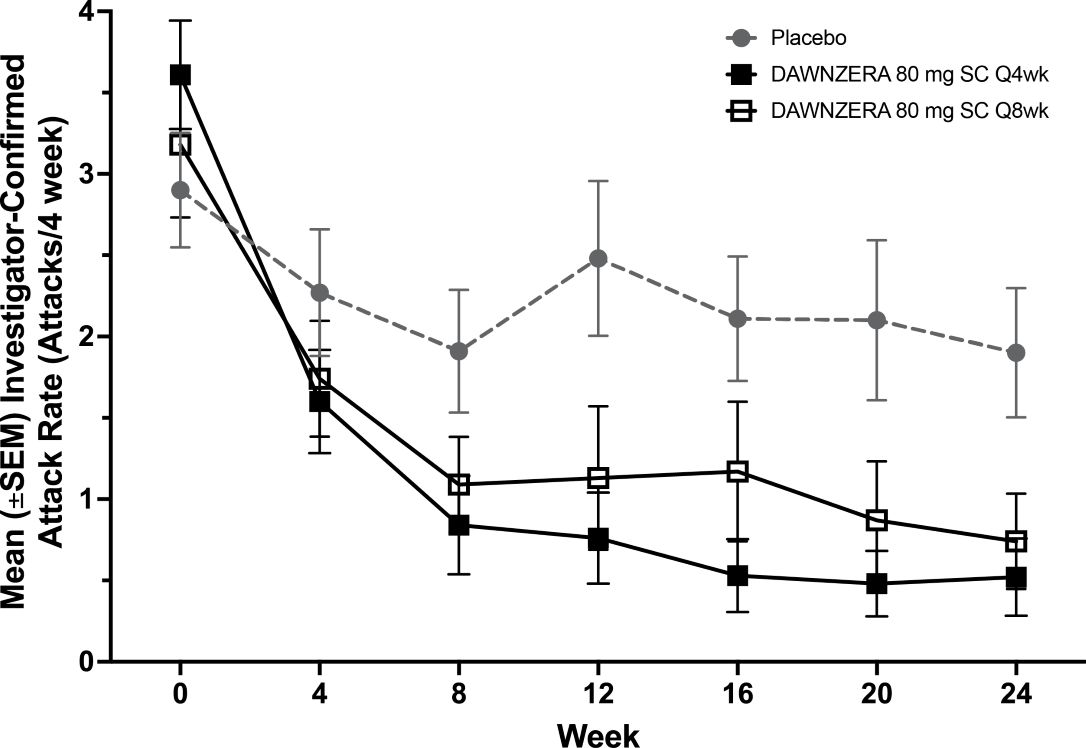
HAE = hereditary angioedema; q4wks = every 4 weeks; q8wks = every 8 weeks; SC = subcutaneous; SEM = standard error of the mean.
Pre-defined secondary endpoints were assessed from Week 4 to Week 24. The moderate or severe HAE attack rate was 0.12 for the DAWNZERA 80 mg every 4 weeks group, 0.68 for the DAWNZERA 80 mg every 8 weeks group, and 1.15 for the placebo group, representing a reduction of 89% (95% CI: 66, 97) and 41% (95% CI: -26, 72) in moderate or severe HAE attack rate relative to placebo, respectively. The HAE attacks requiring acute therapy was 0.15 for the DAWNZERA 80 mg every 4 weeks group, 0.59 for the DAWNZERA 80 mg every 8 weeks group, and 1.80 for the placebo group, representing a reduction of 92% (95% CI: 77, 97) and 67% (95% CI: 29, 85) in HAE attacks requiring acute therapy relative to placebo, respectively.
The proportion of patients who were attack-free from Week 4 to Week 24 were 53% in the DAWNZERA 80 mg every 4 weeks group, 35% in the DAWNZERA 80 mg every 8 weeks group, and 9% in the placebo group, representing an odds ratio of being attack-free of 11.79 (95% CI: 2.34, 59.36) and 3.23 (95% CI: 0.46, 22.85), respectively.
The proportion of patients with a ≥50%, ≥70%, and ≥90% reduction from baseline to Week 4 through Week 24 was 93%, 82%, and 62% in the DAWNZERA 80 mg every 4 weeks group, 83%, 65%, and 48% in the DAWNZERA 80 mg every 8 weeks group, and 27%, 18%, and 9% in the placebo group, respectively.
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
DAWNZERA (donidalorsen) 80 mg/0.8 mL injection is a sterile, preservative‑free, clear, colorless to yellow solution supplied in a single‑dose autoinjector. Each autoinjector of DAWNZERA is filled to deliver 0.8 mL of solution containing 80 mg of donidalorsen. Table 5 provides the presentation and strength for DAWNZERA.
Storage and Handling
- Store the DAWNZERA autoinjector in the refrigerator between 36°F to 46°F (2°C to 8°C) in the original carton.
- The DAWNZERA autoinjector can be stored at room temperature up to 86°F (30°C) in the original carton for up to 6 weeks; if not used within the 6 weeks stored at room temperature, discard DAWNZERA.
- Do not freeze. Do not expose to heat. Protect from direct light.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA‑approved patient labeling (Patient Information and Instructions for Use).
Administration Instructions
- Instruct patients that DAWNZERA is for subcutaneous use and intended for self-administration or administration by a caregiver.
- Instruct patients and/or caregivers on proper preparation and subcutaneous administration technique of DAWNZERA autoinjector [see Dosage and Administration (2.2)and Instructions for Use] .
- Instruct patients to administer DAWNZERA subcutaneously into the abdomen or upper thigh region for self-administration. The back of the upper arm can be used as an injection site if a caregiver or healthcare provider administers DAWNZERA.
Missed Dose(s)
Instruct patients to use DAWNZERA as prescribed. If a dose is missed, instruct patients to administer DAWNZERA as soon as they remember. Instruct patients to resume treatment at the recommended dosing frequency (every 4 weeks or every 8 weeks) from the date of the most recently administered dose [see Dosage and Administration (2.1)] .
Risk of Hypersensitivity Reactions, Including Anaphylaxis
Advise patients hypersensitivity reactions, including anaphylaxis, have been reported following administration of DAWNZERA. Instruct patients to immediately discontinue DAWNZERA and seek medical attention if they experience signs and symptoms of serious hypersensitivity reaction [see Warnings and Precautions (5.1)] .
Distributed by: Ionis Pharmaceuticals Inc., Carlsbad, CA 92010
DAWNZERA is a trademark of Ionis Pharmaceuticals Inc. All other trademarks are the property of their respective owners.
©2025 Ionis Pharmaceuticals Inc.
Instructions for Use

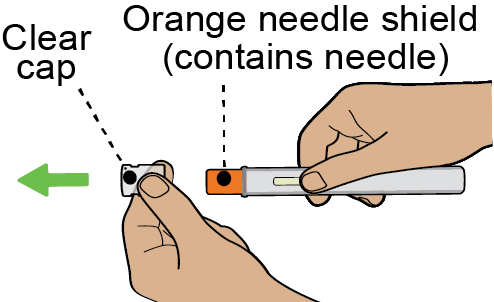
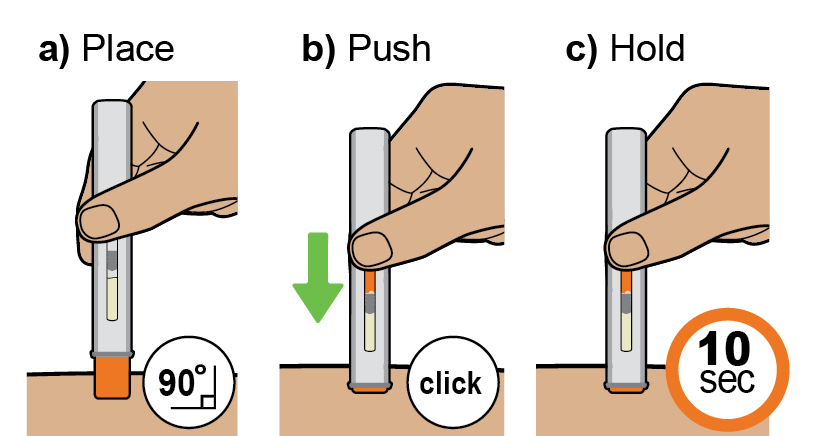
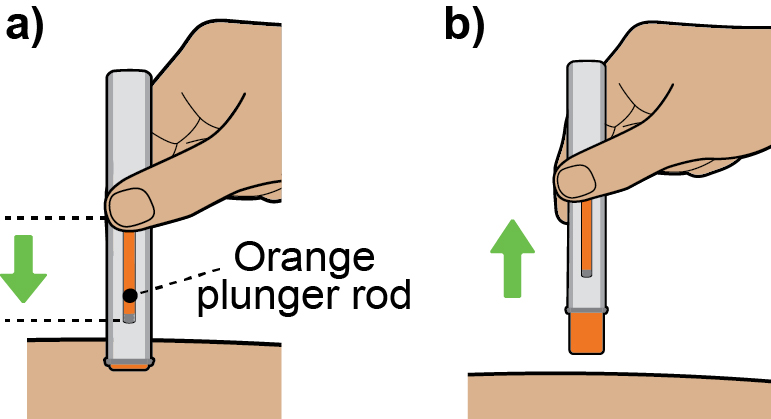
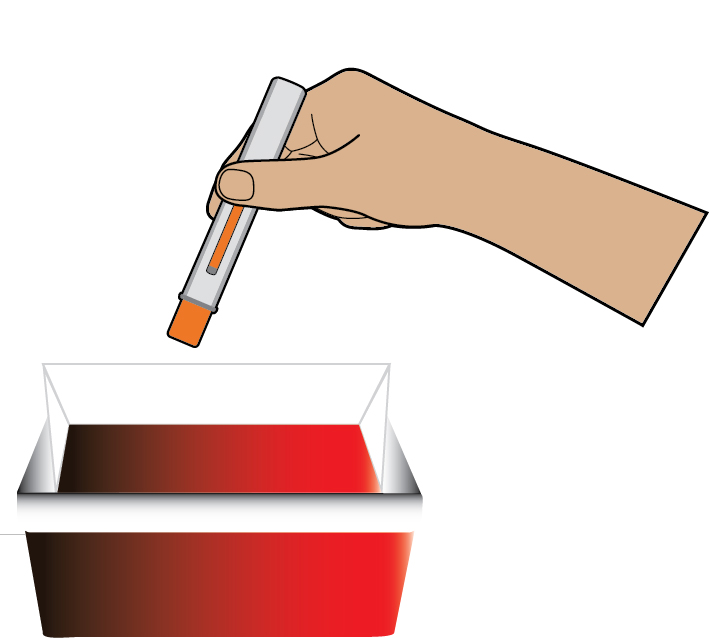
This Instructions for Use has been approved by the U.S. Food and Drug Administration Approved: 08/2025
Patient Information
This Patient Information has been approved by the U.S. Food and Drug Administration Issued: 08/2025
PRINCIPAL DISPLAY PANEL
DAWNZERA™
(donidalorsen) 80mg/0.8mL
injection for subcutaneous use
1 single-dose autoinjector
Each Dawnzera autoinjector contains 80 mg donidalorsen (equivalent to 84 mg donidalorsen
sodium) in 0.8 mL of solution, single-dose, and is for the treatment of adult and adolescent
patients with HAE.
Store refrigerated at 2ºC to 8ºC (36ºC to 46ºC) in the orignal container and protect from direct light.
Do not freeze.
IONIS

Rx Only DAWNZERA™(donidalorsen) 80 mg/0.8ml
injection for subcutaneous use
Each Dawnzera autoinjector contains 80 mg donidalorsen
(equivalent to 84 mg donialorsen sodium)
Store refrigerated at 2ºC to 8ºC (36ºC to 46ºC)
in the original container
Distributed by: NDC71860-103-01
Ionis Pharmaceuticals Inc.
2855 Gazelle Ct, Carlsbad, CA 92010,
United States
IONIS

Guideline Central and select third party use “cookies” on this website to enhance the user experience.
This technology helps us gather statistical and analytical information to optimize the relevant content for you.
The user also has the option to opt-out which may have an effect on the browsing experience.