SOGROYA (somapacitan-beco) injection, solution
1 INDICATIONS AND USAGE
SOGROYA is indicated for the treatment of pediatric patients aged 2.5 years and older with:
-
Growth failure due to inadequate secretion of endogenous growth hormone (GH). -
Short stature born small for gestational age (SGA) and with no catch-up growth by 2 years of age. -
Growth failure associated with Noonan syndrome (NS). -
Idiopathic Short Stature (ISS).
SOGROYA is indicated for the replacement of endogenous GH in adults with growth hormone deficiency (GHD).
SOGROYA is a human growth hormone analog indicated for:
Pediatric Patients: Treatment of pediatric patients aged 2.5 years and older with:
-
Growth failure due to inadequate secretion of endogenous growth hormone (GH). (1) -
Short stature born small for gestational age (SGA) and with no catch-up growth by 2 years of age. (1) -
Growth failure associated with Noonan syndrome (NS). (1) -
Idiopathic Short Stature (ISS). (1)
Adults: Replacement of endogenous growth hormone in adults with growth hormone deficiency (GHD). (1)
2 DOSAGE AND ADMINISTRATION
-
SOGROYA treatment should be supervised by a healthcare provider who is experienced in the diagnosis and management of patients with growth hormone deficiency, pediatric patients born SGA, pediatric patients with NS, and pediatric patients with ISS. (2.1) -
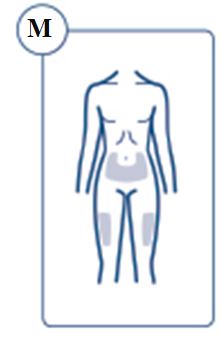
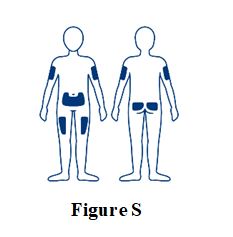
SOGROYA should be administered by subcutaneous injection once weekly, any time of the day, in the upper arms, thigh, abdomen or buttocks with regular rotation of injection site to avoid lipohypertrophy/lipoatrophy. (2.1) -
See Full Prescribing Information for complete dosage, titration, and monitoring recommendations for pediatric and adult patients, including those aged 65 years and older, patients with hepatic impairment, and women receiving oral estrogen. (2.1, 2.2, 2.3, 2.4, 2.5)
For pediatric patients:
-
GH deficiency: Initiate SOGROYA with a dosage of 0.16 mg/kg body weight once weekly for treatment-naïve patients and patients switching from daily growth hormone (somatropin). (2.3) -
SGA/NS/ISS: Initiate SOGROYA with a dosage of 0.24 mg/kg body weight once weekly for treatment-naïve patients and patients switching from daily growth hormone (somatropin). (2.3) -
Individualize dosage for each patient based on the growth response. (2.3) -
Patients switching from daily human growth hormone to once-weekly SOGROYA should choose the preferred day for the weekly dose and stop final dose of daily treatment the day before (or at least 8 hours before) taking the first dose of once-weekly somapacitan-beco. (2.3)
For adult patients with GHD:
-
Initiate SOGROYA with a dosage of 1.5 mg once weekly for treatment naïve patients and patients switching from daily growth hormone. (2.4) -
Increase the weekly dosage every 2 to 4 weeks by approximately 0.5 mg to 1.5 mg until the desired response has been achieved. (2.4) -
Titrate the dosage based on clinical response and serum insulin-like growth factor-1 (IGF-1) concentrations. (2.4) -
The maximum recommended dosage for adult GHD is 8 mg once weekly. (2.4)
2.1 Important Dosing and Administration Information
-
SOGROYA treatment should be supervised by a healthcare provider who is experienced in the diagnosis and management of pediatric patients with growth failure due to GHD, pediatric patients born SGA, pediatric patients with NS, pediatric patients with ISS and/or adults with GHD [see Indications and Usage (1)]. -
SOGROYA should be administered by subcutaneous injection, once weekly, any time of the day, in the upper arms, thigh, abdomen or buttocks with weekly rotation of injection site. -
Inspect visually for particulate matter and discoloration. SOGROYA should be a clear to slightly opalescent and colorless to slightly yellow solution. If the solution is cloudy or contains particulate matter do not use. -
Advise patients to read the PATIENT INFORMATION and INSTRUCTIONS FOR USE leaflets enclosed with the SOGROYA prefilled pen. -
SOGROYA is available in 3 single-patient-use prefilled pens with 3 different dosing ranges (Table 1).
Table 1. Strength and Dosing Range of SOGROYA Prefilled Pens
2.2 Perform Fundoscopic Examination Prior to Initiation of SOGROYA
-
Perform fundoscopic examination before initiating treatment with SOGROYA to exclude preexisting papilledema. If papilledema is identified, evaluate the etiology and treat the underlying cause before initiating treatment with SOGROYA [see Warnings and Precautions (5.5)].
2.3 Recommended Dosage and Monitoring for Pediatric Patients
-
GH Deficiency: Recommended dosage of SOGROYA is 0.16 mg/kg based on actual body weight once weekly for treatment-naïve patients and patients switching from daily growth hormone (somatropin). -
Small for Gestational Age (SGA), Noonan Syndrome (NS), and Idiopathic Short Stature (ISS): Recommended dosage of SOGROYA is 0.24 mg/kg based on actual body weight once weekly for treatment-naïve patients and patients switching from daily growth hormone (somatropin). -
Individualize dosage for each patient based on the growth response. -
When switching from daily human growth hormone to once-weekly SOGROYA, choose the preferred day for the weekly dose. Take the final dose of daily treatment on the day before (or at least 8 hours before) the first dose of SOGROYA. -
When switching from a weekly human growth hormone to once-weekly SOGROYA, continue once weekly dosing schedule. -
Assess compliance and evaluate other causes of poor growth such as hypothyroidism, undernutrition, advanced bone age, and antibodies to recombinant human growth hormone if patients experience failure to increase height velocity, particularly during the first year of treatment. -
Patients who were treated with SOGROYA for GH deficiency in childhood and whose epiphyses are closed should be reevaluated before continuing SOGROYA.
2.4 Recommended Dosage, Titration, and Monitoring for Adults with GHD
-
Initiate SOGROYA with a dosage of 1.5 mg once weekly for treatment naïve patients and patients switching from daily growth hormone (somatropin). -
Increase the weekly dosage every 2 to 4 weeks by approximately 0.5 mg to 1.5 mg until the desired response is achieved. -
Titrate the dosage based on clinical response and serum insulin-like growth factor-1 (IGF-1) concentrations. Draw IGF-1 samples 3 to 4 days after the prior dose. -
Decrease the dosage as necessary on the basis of adverse reactions and/or serum IGF-1 concentrations above the age- and sex-specific normal range. -
The maximum recommended dosage is 8 mg once weekly.
2.5 Recommended Dosage and Titration for Specific Populations
Patients Aged 65 Years and Older
Initiate SOGROYA with a dosage of 1 mg once weekly and use smaller dose increment increases when titrating the dosage [see Use in Specific Populations (8.5)]. See above for monitoring recommendations and the maximum recommended dosage of SOGROYA [see Dosage and Administration (2.4)].
Patients with Hepatic Impairment
-
SOGROYA is not recommended in adult and pediatric patients with severe hepatic impairment [see Hepatic Impairment (8.6)]. -
For adult patients with moderate hepatic impairment, initiate SOGROYA with a dosage of 1 mg once weekly and use smaller dose increment increases when titrating the dosage. See above for monitoring recommendations [see Dosage and Administration (2.4), Pharmacokinetics (12.3)]. The maximum recommended dosage is 4 mg once weekly. -
For pediatric patients with moderate hepatic impairment, SOGROYA is not recommended [see Hepatic Impairment (8.6)]. -
No dosage adjustment is recommended for adult and pediatric patients with mild hepatic impairment.
Women Receiving Oral Estrogen
Initiate SOGROYA with a dosage of 2 mg once weekly [see Drug Interactions (7)]. See above for titration and monitoring recommendations and the maximum recommended dosage of SOGROYA [see Dosage and Administration (2.4)].
2.6 Missed Doses
-
If the dose is missed, SOGROYA can be taken within 3 days after the scheduled dosing day. Once-weekly dosing for the next dose could be resumed at the regularly scheduled dosing day. -
If more than 3 days have passed since the missed dose, skip the dose and administer the next dose on the regularly scheduled dosing day.
3 DOSAGE FORMS AND STRENGTHS
SOGROYA is a clear to slightly opalescent and colorless to slightly yellow solution available as follows:
-
Injection: 5 mg/1.5 mL (3.3 mg/mL) in a single-patient-use prefilled pen (teal) -
Injection: 10 mg/1.5 mL (6.7 mg/mL) in a single-patient-use prefilled pen (yellow) -
Injection: 15 mg/1.5 mL (10 mg/mL) in a single-patient-use prefilled pen (red)
SOGROYA is a liquid solution available in a ready-to-use prefilled pen.
Injection: 5 mg/1.5 mL (3.3 mg/mL) or 10 mg/1.5 mL (6.7 mg/mL) or 15 mg/1.5 mL (10 mg/mL) in a single-patient-use prefilled pen. (3)
4 CONTRAINDICATIONS
SOGROYA is contraindicated in patients with:
-
Acute critical illness after open-heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure because of the risk of increased mortality with use of pharmacologic doses of SOGROYA [see Warnings and Precautions (5.1)]. -
Hypersensitivity to SOGROYA or any of its excipients. Systemic hypersensitivity reactions have been reported postmarketing with somatropin [see Warnings and Precautions (5.2)]. -
Pediatric patients with closed epiphyses. -
Active malignancy [see Warnings and Precautions (5.3)]. -
Active proliferative or severe non-proliferative diabetic retinopathy. -
Pediatric patients with Prader-Willi syndrome who are severely obese, have a history of upper airway obstruction or sleep apnea or have severe respiratory impairment due to risk of sudden death [see Warnings and Precautions (5.13)].
-
Acute critical illness (4) -
Active malignancy (4) -
Hypersensitivity to somapacitan-beco or excipients (4) -
Active proliferative or severe non-proliferative diabetic retinopathy (4) -
Closed epiphyses in children used for longitudinal growth promotion (4) -
Children with Prader-Willi syndrome who are severely obese or have severe respiratory impairment due to risk of sudden death (4)
5 WARNINGS AND PRECAUTIONS
-
Severe Hypersensitivity: Serious hypersensitivity reactions, including anaphylactic reactions and angioedema, may occur. In the event of an allergic reaction, seek prompt medical attention. (5.2) -
Increased Risk of Neoplasm: Monitor patients with preexisting tumors for progression or recurrence. Increased risk of a second neoplasm in childhood cancer survivors treated with somatropin – in particular meningiomas in patients treated with radiation to the head for their first neoplasm. (5.3) -
Glucose Intolerance and Diabetes Mellitus: SOGROYA may decrease insulin sensitivity, particularly at higher doses. Monitor glucose levels periodically in all patients receiving SOGROYA, especially in patients with existing diabetes mellitus or at risk for its development. (5.4) -
Intracranial Hypertension (IH): Perform fundoscopic examinations prior to initiation and periodically thereafter. If papilledema is identified prior to initiation, evaluate the etiology and treat the underlying cause before initiating. If papilledema occurs with SOGROYA, stop treatment. (5.5) -
Fluid Retention: Was observed and may be dose dependent. Reduce dose as necessary. (5.6) -
Hypoadrenalism: Monitor patients for reduced serum cortisol levels and/or need for glucocorticoid dose increases in those with known hypoadrenalism. (5.7) -
Hypothyroidism: Monitor thyroid function periodically as hypothyroidism may occur or worsen after initiation of SOGROYA. (5.8) -
Slipped Capital Femoral Epiphysis in Pediatric Patients: May develop. Evaluate children with the onset of a limp or persistent hip/knee pain. (5.9) -
Progression of Preexisting Scoliosis in Pediatric Patients: May develop. (5.10) -
Pancreatitis: Consider pancreatitis in patients with persistent severe abdominal pain. (5.11) -
Lipohypertrophy/lipoatrophy: May occur if SOGROYA is administered in the same location over a long period of time. Rotate injection sites on a regular basis. (5.12)
5.1 Increased Mortality in Patients with Acute Critical Illness
Increased mortality has been reported after treatment with somatropin in patients with acute critical illness due to complications following open-heart surgery, abdominal surgery and multiple accidental trauma, as well as patients with acute respiratory failure [see Contraindications (4)]. The safety of continuing SOGROYA treatment in patients receiving replacement doses for approved indications who concurrently develop these illnesses has not been established. SOGROYA is not indicated for the treatment of non-GH deficient adults.
5.2 Severe Hypersensitivity
Serious systemic hypersensitivity reactions including anaphylactic reactions and angioedema have been reported with postmarketing use of somatropin. Inform patients and/or caregivers that such reactions are possible, and that prompt medical attention should be sought if an allergic reaction occurs. SOGROYA is contraindicated in patients with known hypersensitivity to somatropin or any excipients in SOGROYA [see Contraindications (4)].
5.3 Increased Risk of Neoplasms
Active Malignancy
There is an increased risk of malignancy progression with somatropin treatment in patients with active malignancy [see Contraindications (4)]. Any preexisting malignancy should be inactive, and its treatment complete prior to instituting therapy with SOGROYA. Discontinue SOGROYA if there is evidence of recurrent activity.
Risk of Second Neoplasm in Pediatric Patients
In childhood cancer survivors who were treated with radiation to the brain/head for their first neoplasm and who developed subsequent growth hormone deficiency (GHD) and were treated with somatropin, an increased risk of second neoplasm has been reported. Intracranial tumors, in particular meningiomas, were the most common of these second neoplasms. Monitor all patients with a history of GHD secondary to an intracranial neoplasm while on somatropin therapy for progression or recurrence of the tumor.
New Malignancy During Treatment
Because children with certain rare genetic causes of short stature have an increased risk of developing malignancies, thoroughly consider the risks and benefits of starting SOGROYA in these patients. If treatment with SOGROYA is initiated, carefully monitor these patients for development of neoplasms.
There is risk of malignant changes of preexisting nevi with somatropin treatment in patients. Monitor patients on SOGROYA therapy carefully for increased growth or potential malignant changes of preexisting nevi. Advise patients/caregivers to report marked changes in behavior, onset of headaches, vision disturbances, and/or changes in the appearance of preexisting nevi.
5.4 Glucose Intolerance and Diabetes Mellitus
Treatment with somatropin may decrease insulin sensitivity, particularly at higher doses. New onset type 2 diabetes mellitus has been reported in patients taking somatropin. Patients with undiagnosed pre-diabetes and diabetes mellitus may experience worsened glycemic control and become symptomatic. Monitor glucose levels periodically in all patients receiving SOGROYA, especially in those with risk factors for diabetes mellitus, such as obesity, Turner syndrome, or a family history of diabetes mellitus. Patients with preexisting type 1 or type 2 diabetes mellitus or pre-diabetes should be monitored closely. The doses of antidiabetic agents may require adjustment when SOGROYA is initiated.
5.5 Intracranial Hypertension
Intracranial hypertension with papilledema, visual changes, headache, nausea, and/or vomiting has been reported in patients treated with somatropin. Symptoms usually occurred within the first eight (8) weeks after the initiation of somatropin therapy. In all reported cases, intracranial hypertension-associated signs and symptoms rapidly resolved after cessation of therapy or a reduction of the somatropin dose.
Perform fundoscopic examination before initiating treatment with SOGROYA to exclude preexisting papilledema and periodically thereafter. If papilledema is identified prior to initiation, evaluate the etiology and treat the underlying cause before initiating SOGROYA. If papilledema is observed by fundoscopy during SOGROYA treatment, treatment should be stopped. If intracranial hypertension is confirmed, treatment with SOGROYA can be restarted at a lower dose after intracranial hypertension-associated signs and symptoms have resolved.
5.6 Fluid Retention
Fluid retention was observed during SOGROYA therapy. Clinical manifestations of fluid retention (e.g. edema and nerve compression syndromes including carpal tunnel syndrome/paresthesia) are usually transient and dose dependent.
5.7 Hypoadrenalism
Patients receiving somatropin therapy who have or are at risk for corticotropin deficiency may be at risk for reduced serum cortisol levels and/or unmasking of central (secondary) hypoadrenalism. In addition, patients treated with glucocorticoid replacement for previously diagnosed hypoadrenalism may require an increase in their maintenance or stress doses following initiation of SOGROYA treatment. Monitor patients with known hypoadrenalism for reduced serum cortisol levels and/or need for glucocorticoid dose increases [see Drug Interactions (7)].
5.8 Hypothyroidism
Undiagnosed/untreated hypothyroidism may prevent an optimal response to SOGROYA. In patients with GH deficiency, central (secondary) hypothyroidism may first become evident or worsen during treatment with somatropin therapy. Therefore, patients should have periodic thyroid function tests and thyroid hormone replacement therapy should be initiated or appropriately adjusted when indicated.
5.9 Slipped Capital Femoral Epiphysis in Pediatric Patients
Slipped capital femoral epiphysis may occur more frequently in patients undergoing rapid growth. Slipped capital femoral epiphysis may lead to osteonecrosis. Cases of slipped capital femoral epiphysis with or without osteonecrosis have been reported in pediatric patients with short stature receiving somatropin. Evaluate pediatric patients receiving SOGROYA with the onset of a limp or complaints of persistent hip or knee pain for slipped capital femoral epiphysis and osteonecrosis and manage accordingly.
5.10 Progression of Preexisting Scoliosis in Pediatric Patients
Somatropin, including SOGROYA, increases growth rate, and progression of preexisting scoliosis can occur in patients who experience rapid growth. Somatropin has not been shown to increase the occurrence of scoliosis. Monitor patients with a history of scoliosis for disease progression.
5.11 Pancreatitis
Cases of pancreatitis have been reported in patients receiving somatropin. The risk may be greater in pediatric patients compared with adults. Consider pancreatitis in patients who develop persistent severe abdominal pain.
5.12 Lipohypertrophy/Lipoatrophy
When SOGROYA is administered subcutaneously at the same site over a long period of time, tissue lipohypertrophy or lipoatrophy may result. Rotate injection sites when administering SOGROYA to reduce this risk [see Dosage and Administration (2.1)].
5.13 Sudden Death in Pediatric Patients with Prader-Willi Syndrome
There have been reports of fatalities after initiating therapy with somatropin in pediatric patients with Prader-Willi syndrome who had one or more of the following risk factors: severe obesity, history of upper airway obstruction or sleep apnea, or unidentified respiratory infection. Male patients with one or more of these factors may be at greater risk than females. SOGROYA is not indicated for the treatment of pediatric patients who have growth failure due to genetically confirmed Prader-Willi syndrome.
5.14 Laboratory Tests
Serum levels of inorganic phosphorus and alkaline phosphatase have increased after somatropin therapy, including SOGROYA. Serum levels of parathyroid hormone may increase with somatropin treatment.
6 ADVERSE REACTIONS
The following clinically significant adverse drug reactions are described elsewhere in the labeling:
-
Increased mortality in patients with acute critical illness [see Warnings and Precautions (5.1)] -
Severe hypersensitivity [see Warnings and Precautions (5.2)] -
Increased risk of neoplasms [see Warnings and Precautions (5.3)] -
Glucose intolerance and diabetes mellitus [see Warnings and Precautions (5.4)] -
Intracranial hypertension [see Warnings and Precautions (5.5)] -
Fluid retention [see Warnings and Precautions (5.6)] -
Hypoadrenalism [see Warnings and Precautions (5.7)] -
Hypothyroidism [see Warnings and Precautions (5.8)] -
Slipped capital femoral epiphysis in pediatric patients [see Warnings and Precautions (5.9)] -
Progression of preexisting scoliosis in pediatric patients [see Warnings and Precautions (5.10)] -
Pancreatitis [see Warnings and Precautions (5.11)] -
Lipohypertrophy/Lipoatrophy [see Warnings and Precautions (5.12)] -
Sudden death in pediatric patients with Prader-Willi syndrome [see Warnings and Precautions (5.13)]
-
Common adverse reactions reported in pediatric patients treated with SOGROYA include: cough, diarrhea, ear infection, headache, injection site reaction, nasopharyngitis, pain in extremity, pyrexia, respiratory tract infection, and vomiting. (6.1) -
Adult patients with GHD: Adverse reactions reported in >2% of patients treated with SOGROYA are: back pain, arthralgia, dyspepsia, sleep disorder, dizziness, tonsillitis, peripheral edema, vomiting, adrenal insufficiency, hypertension, blood creatine phosphokinase increase, weight increase, anemia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novo Nordisk Inc. at 1-800-727-6500 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Pediatric Patients with GHD
SOGROYA 0.16 mg/kg/week was studied in a 52-week randomized, open-label, active-controlled, parallel-group clinical study in 200 treatment-naïve, prepubertal pediatric patients with growth hormone deficiency [see Clinical Studies (14.1)]. Table 2 shows common adverse reactions that occurred in ≥5% of patients treated with either SOGROYA or somatropin in this trial.
Table 2. Adverse Reactions Occurring ≥5% in SOGROYA or Somatropin-treated Pediatric Patients (52 Weeks of Treatment)
-
-
-
-
-
-
Pediatric Patients Born Small for Gestational Age and with no Catch-up Growth by 2 Years of Age
SOGROYA 0.24 mg/kg/week was studied in a 52-week randomized, open-label, active-comparator, basket clinical study in GH treatment-naïve, pre-pubertal patients with short stature born small for gestational age (SGA), Noonan syndrome (NS) or idiopathic short stature (ISS) compared to somatropin [see Clinical Studies (14.2)]. Table 3 shows the adverse reactions occurring at ≥10% in the SGA cohort with 141 pediatric patients.
Table 3. Adverse Reactions Occurring ≥10% in SOGROYA or Somatropin-Treated Pediatric Patients with SGA at Week 52
-
-
c Cough in the SOGROYA treatment group included cough (15.9%).
d Ear infection in the SOGROYA treatment group included otitis media (7.2%), ear infection (2.9%), otitis externa (1.4%), and otitis media acute (1.4%).
e Diarrhea in the SOGROYA treatment group included diarrhea (4.3%), gastroenteritis (4.3%), and gastrointestinal viral infection (1.4%)
f Vomiting in the SOGROYA treatment group included vomiting (10.1%)
Description of Select Adverse Reactions
Adenoidal and tonsillar hypertrophy was reported in 1 (1%) subject with SGA in the first year of treatment with SOGROYA who received tonsillectomy and adenoidectomy. The adenoidal and tonsillar hypertrophy resolved after the procedure.
Pediatric Patients with Growth Failure Associated with Noonan Syndrome
SOGROYA 0.24 mg/kg/week was studied in a 52-week randomized, open-label, active-comparator, basket clinical study in GH treatment-naïve, pre-pubertal patients with NS compared to somatropin 0.05 mg/kg/day [see Clinical Studies (14.3)]. Table 4 shows the adverse reactions occurring at ≥10% in the NS cohort with 77 pediatric patients.
Table 4. Adverse Reactions Occurring ≥10% in SOGROYA or Somatropin-Treated Pediatric Patients with NS at Week 52
a Respiratory tract infection in the SOGROYA treatment group included upper respiratory tract infection (20.4%), influenza (10.2%), pneumonia (6.1%), influenza like illness (2%), respiratory syncytial virus infection (2%), respiratory tract infection (2%), respiratory tract infection viral (2%), upper respiratory tract infection bacterial (2%), and viral upper respiratory tract infection (2%).
b Nasopharyngitis in the SOGROYA treatment group included nasopharyngitis (24.5%), pharyngitis (4.1%), bacterial infection (2%), herpes pharyngitis (2%), pharyngitis streptococcal (2%), pharyngotonsillitis (2%), and rhinitis (2%).
c Diarrhea in the SOGROYA treatment group included gastroenteritis (14.3%), diarrhea (6.1%), gastroenteritis viral (2%), and parasitic gastroenteritis (2%).
d Ear infection in the SOGROYA treatment group included otitis media (8.2%), otitis externa (4.1%), ear infection (2%), otitis media acute (2%), and otitis media chronic (2%).
e Cough in the SOGROYA treatment group included cough (12.2%) and bacterial infection (2%).
f Vomiting in the SOGROYA treatment group included vomiting (8.2%) and gastritis (4.1%).
g Injection site reaction in the SOGROYA treatment group included injection site bruising (6.1%) and injection site hemorrhage (2%)
h Abdominal pain in the SOGROYA treatment group included abdominal distension (2%)
Pediatric Patients with Growth Failure Associated with Idiopathic Short Stature
SOGROYA 0.24 mg/kg/week was studied in a 52-week randomized, open-label, active-comparator, basket clinical study in GH treatment-naïve, pre-pubertal patients with ISS compared to somatropin 0.05 mg/kg/day [see Clinical Studies (14.4)]. Table 5 shows the adverse reactions occurring at ≥10% in the ISS cohort with 87 pediatric patients.
Table 5. Adverse Reactions Occurring ≥10% in SOGROYA or Somatropin-Treated Pediatric Patients with ISS at Week 52
a Respiratory tract infection in the SOGROYA treatment group included influenza (16.9%), upper respiratory tract infection (6.8%), respiratory tract infection (3.4%), pneumonia (1.7%), pneumonia bacterial (1.7%), respiratory tract infection viral (1.7%), and viral upper respiratory tract infection (1.7%).
b Nasopharyngitis in the SOGROYA treatment group included nasopharyngitis (15.3%), pharyngitis streptococcal (5.1%), pharyngitis (1.7%), rhinitis (1.7%), sinusitis (1.7%), and tracheitis (1.7%).
c Ear infection in the SOGROYA treatment group included otitis media (6.8%), ear infection (3.4%), and otitis media acute (1.7%).
d Diarrhea in the SOGROYA treatment group included gastroenteritis (3.4%), gastroenteritis viral (3.4%), diarrhea (1.7%), enterobiasis (1.7%), and parasitic gastroenteritis (1.7%).
e Injection site reaction in the SOGROYA treatment group included application site reaction (1.7%), injection site bruising (1.7%), injection site hematoma (1.7%), injection site hemorrhage (1.7%), injection site pruritis (1.7%), and injection site urticaria (1.7%).
f Cough in the SOGROYA treatment group included cough (5.1%).
g Vomiting in the SOGROYA treatment group included vomiting (5.1%).
Description of Select Adverse Reactions
Adenoidal and tonsillar hypertrophy was reported in 1 (2%) subject with ISS in the first year of treatment with SOGROYA who received tonsillectomy and adenoidectomy. The adenoidal and tonsillar hypertrophy resolved after the procedure.
Adult Patients with GHD
SOGROYA was studied in adult patients with GHD in a 35-week, placebo-controlled, double-blind trial with an active-control arm [see Clinical Studies (14.5)]. Adverse reactions occurring >2% with SOGROYA are presented in Table 6.
Table 6. Adverse Reactions Occurring >2% in Adults with GHD Treated with SOGROYA and More Frequently# than in Placebo-Treated Patients for 34 Weeks
-
More SOGROYA treated patients shifted from normal baseline levels to elevated phosphate and creatine phosphokinase levels at the end of the trial compared to the placebo group (17.5% vs 4.9% and 9.2% vs. 6.6%, respectively); these laboratory changes occurred intermittently, and were non-progressive.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of somatropins. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Musculoskeletal and connective tissue disorders – osteonecrosis in pediatric patients.
7 DRUG INTERACTIONS
Table 7 includes a list of drugs with clinically important drug interactions when administered concomitantly with SOGROYA and instructions for preventing or managing them.
Table 7. Clinically Important Drug Interactions with SOGROYA
-
Replacement Glucocorticoid Treatment: Patients treated with glucocorticoid for hypoadrenalism may require an increase in their maintenance or stress doses following initiation of SOGROYA. (7) -
Cytochrome P450-Metabolized Drugs: SOGROYA may alter the clearance. Monitor carefully if used with SOGROYA. (7) -
Oral Estrogen: Larger doses of SOGROYA may be required. (7) -
Insulin and/or Other Antihyperglycemic Agents: Dose adjustment of insulin or antihyperglycemic agent may be required. (5.4, 7)
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on the use of SOGROYA during pregnancy; however, published studies describing the use of short-acting recombinant growth hormone (rhGH) during pregnancy over several decades have not identified any drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction studies, subcutaneously administered somapacitan-beco was not teratogenic in rats or rabbits during organogenesis at doses approximately 12 times the clinical exposure at the maximum recommended human dose (MRHD) of 8 mg/week. No adverse developmental outcomes were observed in a pre- and post-natal development study with administration of somapacitan-beco to pregnant rats from organogenesis through lactation at approximately 275 times the clinical exposure at the MRHD (see Data).
The background risk of birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in rats, somapacitan-beco was administered by subcutaneous injection at doses of 2, 6, and 18 mg/kg/day during the period of organogenesis from gestation day 6 to 17. Fetal viability and development were not affected at doses up to 6 mg/kg/day (31 times the MRHD, based on AUC). Transient, fetal skeletal variations (short/bent/thickened long bones) were observed at 18 mg/kg/day (261 times the MRHD, based on AUC).
In an embryo-fetal development study in rabbits, somapacitan-beco was administered by subcutaneous injection at doses of 1, 3, and 9 mg/kg every two days during the period of organogenesis from gestation day 6 to 18. Fetal viability and development were not adversely affected at somapacitan-beco dose of 1 mg/kg/every two days (12 times the MRHD, based on AUC). Reduced fetal growth was observed at doses ≥3 mg/kg/every two days (≥130 times the MRHD, based on C12h).
In a pre- and post-natal development study in pregnant rats, somapacitan-beco was administered by subcutaneous injection at doses of 4, 9, and 18 mg/kg twice a week from gestation day 6 through lactation day 18. No adverse developmental effects were observed in the offspring at doses up to 9 mg/kg (275 times the MRHD, based on AUC). Increased incidence of renal pelvic dilatation was observed on post-natal day 21 at 18 mg/kg (630 times the MRHD, based on AUC), but was not observed in the adult F1 generation.
8.2 Lactation
Risk Summary
There is no information on the presence of somapacitan-beco in human milk, the effects on the breastfed infant, or the effects on milk production. Somapacitan-beco-related material was secreted into milk of lactating rats. When a substance is present in animal milk, it is likely that the substance will be present in human milk. Available published data describing administration of short-acting recombinant growth hormone (rhGH) to lactating women for 7 days reported that short-acting rhGH did not increase the normal breastmilk concentration of growth hormone and no adverse effects were reported in breastfed infants. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for SOGROYA and any potential adverse effects on the breastfed infant from SOGROYA or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of SOGROYA have been established in pediatric patients 2.5 years of age and older for the treatment of:
-
Growth failure due to inadequate secretion of endogenous GH. The use of SOGROYA for this indication is supported by evidence from a 52-week randomized, multi-center, open-label, active-controlled, parallel-group phase 3 trial in 200 treatment-naïve, pediatric patients aged 2.5 to 11 years with GHD [see Clinical Studies (14.1)]. The safety profile from the pediatric trial was similar to that reported in adults [see Adverse Reactions (6.1)]. -
Short stature born SGA with no catch-up growth by 2 years of age. The use of SOGROYA for this indication is supported by evidence from a multi-center, randomized open-label, active-comparator, phase 3 basket study in 142 pediatric patients aged 2.6 to 10.7 years with short stature born SGA with no catch-up growth by 2 years of age [see Clinical Studies (14.2)]. -
Growth failure associated with NS. The use of SOGROYA for this indication is supported by evidence from a multi-center, randomized open-label, active-comparator, phase 3 basket study in 77 pediatric patients aged 2 to 11.1 years with growth failure associated with NS [see Clinical Studies (14.3)]. -
ISS. The use of SOGROYA for this indication is supported by evidence from a multi-center, randomized open-label, active-comparator, phase 3 basket study in 88 pediatric patients aged 2.8 to 10.8 years with ISS [see Clinical Studies (14.4)].
The safety and effectiveness of SOGROYA have not been established in pediatric patients less than 2.5 years of age for the treatment of growth failure due to inadequate secretion of endogenous GH, short stature born SGA with no catch-up growth, growth failure associated with NS, or with ISS.
Risks in pediatric patients associated with growth hormone use include:
-
Sudden death in pediatric patients with Prader-Willi Syndrome. SOGROYA is not indicated for the treatment of pediatric patients with growth failure secondary to genetically confirmed Prader-Willi syndrome. [see Warnings and Precautions (5.13)] -
Increased risk of second neoplasm in pediatric cancer survivors treated with radiation to the brain and/or head [see Warnings and Precautions (5.3)] -
Slipped capital femoral epiphysis in pediatric patients [see Warnings and Precautions (5.9)] -
Progression of preexisting scoliosis in pediatric patients [see Warnings and Precautions (5.10)] -
Pancreatitis [see Warnings and Precautions (5.11)]
8.5 Geriatric Use
In clinical studies a total of 52 (15.6%) of the 333 SOGROYA-treated patients were 65 years or older and 3 (0.9%) were 75 years or older [see Clinical Studies (14)]. Subjects older than 65 years appeared to have higher exposure than younger subjects at the same dose level. Elderly patients may be more sensitive to the action of somapacitan-beco and therefore may be at increased risk for adverse reactions. Initiate SOGROYA with a dose of 1 mg once weekly and use smaller increments when increasing the dose [see Dosage and Administration (2.5)].
8.6 Hepatic Impairment
Adult patients: No dose adjustment of SOGROYA is required for patients with mild hepatic impairment. Higher somapacitan-beco exposure was observed in patients with moderate hepatic impairment. In patients with moderate hepatic impairment, initiate SOGROYA with a dose of 1 mg once weekly and use smaller increments when increasing the dose. The maximum dose should not exceed 4 mg once weekly. Somapacitan-beco was not studied in patients with severe hepatic impairment. Therefore, use of SOGROYA is not recommended in patients with severe hepatic impairment. [see Dosage and Administration (2.5), Clinical Pharmacology (12.3)].
Pediatric patients: Based on the hepatic impairment study in adults, no dose adjustment of SOGROYA is recommended for patients with mild hepatic impairment. Higher systemic exposure of SOGROYA is expected in pediatric patients with moderate and severe hepatic impairment; therefore, SOGROYA is not recommended in these pediatric patients [see Dosage and Administration (2.5)].
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
SOGROYA contains somapacitan-beco, which is not a controlled substance.
9.2 Abuse
Inappropriate use of SOGROYA may result in significant negative health consequences.
9.3 Dependence
SOGROYA is not associated with drug related withdrawal adverse reactions.
10 OVERDOSAGE
Acute overdosage could lead initially to hypoglycemia and subsequently to hyperglycemia. Overdose with SOGROYA is likely to cause fluid retention. Long-term overdosage could result in signs and symptoms of gigantism and/or acromegaly consistent with the known effects of excess endogenous growth hormone.
11 DESCRIPTION
Somapacitan-beco is a human growth hormone (hGH) analog with a single substitution in the amino acid backbone (L101C) to which an albumin-binding moiety has been attached. The albumin-binding moiety (side-chain) consists of an albumin binder and a hydrophilic spacer attached to position 101 of the protein. The protein part consists of 191 amino acids. Somapacitan-beco is produced in Escherichia coli by recombinant DNA technology. The molecular formula (including the albumin-binding moiety) is C1038H1609N273O319S9 and the molecular weight is 23305.10 g/mol, of which the albumin-binding moiety is 1191.39 g/mol.
Structural Formula:

SOGROYA (somapacitan-beco) injection is supplied as a sterile, clear to slightly opalescent and colorless to slightly yellow solution for subcutaneous use in a single-patient-use prefilled pen with a deliverable volume of 1.5 mL.
Each mL of SOGROYA 5 mg/1.5 mL prefilled pen contains 3.3 mg of somapacitan-beco, histidine (0.68 mg), mannitol (44 mg), phenol (4 mg), poloxamer 188 (1 mg), and Water for Injection, USP. The pH is approximately 6.8. Hydrochloric acid and sodium hydroxide may be added to adjust the pH.
Each mL of SOGROYA 10 mg/1.5 mL prefilled pen contains 6.7 mg of somapacitan-beco, histidine (0.68 mg), mannitol (44 mg), phenol (4 mg), poloxamer 188 (1 mg), and Water for Injection, USP. The pH is approximately 6.8. Hydrochloric acid and sodium hydroxide may be added to adjust the pH.
Each mL of SOGROYA 15 mg/1.5 mL prefilled pen contains 10 mg of somapacitan-beco, histidine (0.68 mg), mannitol (44 mg), phenol (4 mg), poloxamer 188 (1 mg), and Water for Injection, USP. The pH is approximately 6.8. Hydrochloric acid and sodium hydroxide may be added to adjust the pH.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Somapacitan-beco binds to a dimeric GH receptor in the cell membrane of target cells resulting in intracellular signal transduction and a host of pharmacodynamic effects. Some of these pharmacodynamic effects are primarily mediated by insulin-like growth factor-1 (IGF-1) produced in the liver, while others are primarily a consequence of the direct effects of somapacitan-beco.
12.2 Pharmacodynamics
IGF-1 was measured to assess the pharmacodynamic (PD) properties of somapacitan-beco. Somapacitan-beco normalizes the mean IGF-1 standard deviation score (SDS) level from a baseline value below -2 to a value within the reference range (-2 to +2) in treatment-naïve adult patients with GHD [see Clinical Studies (14)].
In adult patients with GHD (n=26), somapacitan-beco induces a less than dose proportional IGF-1 response at steady state. Maximum IGF-1 concentrations were observed within 2 to 4 days after dosing. Similar to the somapacitan-beco exposure time course, a steady state IGF-1 response was reached after 1 to 2 weekly doses with limited cumulative IGF-1 response.
In pediatric patients with GHD aged 2.5 to 11 years, somapacitan-beco produces a dose linear IGF-1 response, with a change of 0.02 mg/kg on average resulting in a change in IGF-1 standard deviation score (SDS) of 0.32. Approximately 97% of pediatric patients achieved an average IGF-1 SDS level within normal range after 52 weeks of treatment with once weekly SOGROYA in Study NCT03811535. IGF-1 SDS levels were -2.03 at baseline and the IGF-1 SDS level change from baseline was 2.36.
In pediatric patients born SGA aged 2.6 to 10.1 years, somapacitan-beco produces a dose linear IGF-1 response, with a change of 0.02 mg/kg/week on average resulting in a change in IGF-1 SDS of 0.24 in the dosing range of 0.16 mg to 0.24 mg/kg/week. IGF-1 SDS was normal at baseline and increased by 2.48 after 52 weeks of treatment with SOGROYA 0.24 mg/kg/week.
In pediatric patients with NS aged 2.6 to 11.1 years, IGF-1 SDS was normal at baseline and increased by 2.35 after 52 weeks of treatment with SOGROYA 0.24 mg/kg/week.
In pediatric patients with ISS aged 2.8 to 10.8 years, IGF-1 SDS was normal at baseline and increased by 2.44 after 52 weeks of treatment with SOGROYA 0.24 mg/kg/week.
12.3 Pharmacokinetics
Somapacitan-beco has pharmacokinetic properties compatible with once weekly administration. The reversible binding to endogenous albumin delays elimination of somapacitan and thereby prolongs the in vivo half-life and duration of action.
The pharmacokinetics (PK) of somapacitan-beco following subcutaneous administration have been investigated at clinically relevant doses (e.g., 0.01 to 0.32 mg/kg in healthy adults, 0.02 to 0.12 mg/kg in adults with GHD, 0.02 to 0.16 mg/kg in pediatric patients with GHD, 0.16 to 0.24 mg/kg in pediatric patient born SGA, 0.24 mg/kg in pediatric patient with NS, and 0.24 mg/kg in pediatric patient with ISS).
Overall, somapacitan-beco displays non-linear pharmacokinetics, however in the clinically relevant dose range of somapacitan-beco in adults with GHD, somapacitan-beco pharmacokinetics are approximately linear. After subcutaneous administration of 0.02 – 0.16 mg/kg/week somapacitan-beco in pediatric patients with GHD, a non-linear dose-exposure relationship with a greater than dose proportional increase in exposure was observed.
Absorption
In adults and pediatric patients, steady state exposure is achieved following 1 to 2 weeks of once weekly administration of subcutaneous somapacitan-beco.
In adults with GHD, a maximum concentration of somapacitan-beco is reached 4 to 24 hours post dose.
In pediatric patients with GHD, maximum somapacitan-beco concentrations occurred 8 to 25 hours after dosing at doses from 0.02 mg/kg/week to 0.16 mg/kg/week and increased with increasing dose level.
In pediatric patients born SGA, a maximum concentration of somapacitan-beco is reached 22 hours post dose at doses from 0.16 mg/kg/week to 0.24 mg/kg/week and increased with increasing dose level.
In pediatric patients with NS, a maximum concentration of somapacitan-beco is reached 23 hours post dose at 0.24 mg/kg/week.
In pediatric patients with ISS, a maximum concentration of somapacitan-beco is reached 23 hours post dose at 0.24 mg/kg/week.
Distribution
Somapacitan-beco is extensively bound (>99%) to plasma proteins.
Based on population PK analyses, the estimated volume of distribution (V/F) of somapacitan-beco in adult GHD patients is approximately 14.6 L, 1.7 L in pediatric patients with GHD, 2.97 L in pediatric patients born SGA, 3.23 L in pediatric patients with NS, and 2.99 L in pediatric patients with ISS.
Elimination
The plasma elimination half-life of somapacitan-beco is approximately 2 to 3 days in adult patients with GHD. Following a dose of 0.16 mg/kg/week, the terminal half-life of somapacitan-beco was about 34 hours in pediatric patients with GHD.
Following a dose of 0.24 mg/kg/week, the mean terminal half-life of somapacitan-beco range from 36 to 37 hours in pediatric patients with SGA, NS, and ISS.
Somapacitan-beco was cleared within one week after treatment discontinuation.
Metabolism: Somapacitan-beco is metabolized via proteolytic cleavage of the linker sequence between the peptide backbone and albumin binder sidechain.
Excretion: The primary excretion routes of somapacitan-beco-related material are via the urine and feces. Approximately 81% of the dose is excreted in the urine and approximately 13% is excreted in the feces. No intact somapacitan-beco is excreted indicating full breakdown of somapacitan-beco prior to excretion.
Specific Populations
Body weight: Adults with GHD – The exposure of somapacitan-beco decreases with increasing body weight. However, the somapacitan-beco dose range of 0.1 to 8 mg/week provides adequate systemic exposure to reach target IGF-1 levels over the weight range of 34.5-150.5 kg evaluated in the clinical trials.
Pediatric patients with GHD: Based on pharmacokinetic analysis, gender and race do not have a clinically meaningful effect on the pharmacokinetics. The exposure of somapacitan-beco decreases with increasing body weight. However, the somapacitan-beco dose of 0.16 mg/kg/week provides adequate systemic exposure for pediatrics to reach target IGF-1 levels over the weight range of 9.9 to 61.8 kg evaluated in the clinical trials.
Geriatric patients: Adult patients greater than 65 years of age and geriatric patients have a higher exposure than younger subjects at the same somapacitan-beco dose [see Dosage and Administration (2.5), Use in Specific Populations (8.5)].
Female patients receiving estrogen: Female patients and in particular female patients on oral estrogen, have lower exposure than males at the same somapacitan-beco dose [see Dosage and Administration (2.5), Drug Interactions (7)].
Hepatic impairment: A somapacitan-beco dose of 0.08 mg/kg at steady state resulted in comparable somapacitan-beco exposure between patients with normal hepatic function and mild hepatic impairment (Child-Pugh A). However, higher exposure was observed in patients with moderate hepatic impairment (Child-Pugh B) (ratios to normal hepatic function were 4.69 and 3.52-fold increase for AUC0-168h and Cmax, respectively). Lower somapacitan-beco stimulated IGF-1 levels were observed in patients with mild and moderate hepatic impairment (ratios to normal hepatic function were 0.85 and 0.75, respectively) [see Dosage and Administration (2.5), Use in Specific Populations (8.6)].
Renal impairment: In general, somapacitan-beco exposure tended to increase with decreasing estimated glomerular filtration rate. A somapacitan-beco dose of 0.08 mg/kg at steady state resulted in higher exposures in patients with renal impairment, that was most pronounced for patients with severe renal impairment and patients requiring hemodialysis (AUC0-168h ratios to normal renal function were 1.75 and 1.63, respectively). Higher IGF-1 AUC0-168h levels were also observed in patients with moderate and severe renal impairment and in patients requiring hemodialysis (ratios to normal renal function were 1.35, 1.40 and 1.24, respectively).
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, include those of SOGROYA or other growth hormone analogs.
No anti-somapacitan-beco-antibodies were detected in the clinical trials in adult patients with GHD.
For pediatric patients with GHD, of the 132 subjects exposed to somapacitan-beco, 16 (12.1%) were ADA positive at any time during the 52-week main period of the trial following exposure to SOGROYA. Fourteen of the 16 subjects showed positive ADA only at one timepoint.
For pediatric patients born SGA, of the 69 subjects exposed to somapacitan-beco, 10 (14.5 %) were ADA positive at any time during the 52-week main period of the trial following exposure to SOGROYA. Three of the 10 subjects showed positive ADA at only one time point.
For pediatric patients with NS, of the 49 subjects exposed to somapacitan-beco, 2 (4.1 %) were ADA positive at any time during the 52-week main period of the trial following exposure to SOGROYA. One of the 2 subjects showed positive ADA at only one time point.
For pediatric patients with ISS, of the 59 subjects exposed to somapacitan-beco, 7 (11.9 %) were ADA positive at any time during the 52-week main period of the trial following exposure to SOGROYA. Three of the 7 subjects showed positive ADA at only one time point.
There was no identified clinically significant effect of ADA on pharmacokinetics, pharmacodynamics, safety, or effectiveness of SOGROYA over the treatment duration. No neutralizing antibodies to somapacitan-beco were detected across the studied pediatric patient populations.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long term studies in animals with somapacitan-beco to evaluate carcinogenic potential have not been conducted.
Somapacitan-beco was not mutagenic or clastogenic in a standard battery of genotoxicity tests (bacterial mutagenicity (Ames), human lymphocyte chromosome aberration, rat bone marrow micronucleus).
In rat studies evaluating male and female fertility, somapacitan-beco was administered by subcutaneous injection at doses of 1, 2, and 4 mg/kg twice weekly. Males were dosed from four weeks before pairing until termination and females were dosed beginning two weeks prior to mating through gestation day 7. No adverse effects were observed on male or female fertility in rats at doses up to 4 mg/kg (29 times the MRHD, based on AUC).
14 CLINICAL STUDIES
14.1 Pediatric Patients with Growth Hormone Deficiency (GHD)
A randomized, open-label, active-controlled, parallel-group phase 3 study was conducted in 200 treatment-naïve, pediatric patients with growth hormone deficiency (GHD) (NCT03811535). The primary efficacy endpoint was annualized height velocity at Week 52.
One hundred thirty-two patients (132) received 0.16 mg/kg/week SOGROYA, and 68 received 0.034 mg/kg/day daily somatropin. The patients ranged in age from 2.5 to 11 years with a mean of 6.4 years. Of these patients, 74.5% were male and 25.5% were female. Fifty-seven percent (57%) of patients were Caucasian, 37% of patients were Asian, 0.5% of patients were Black or African American, 5.0% were not reported, and 0.5% were categorized as “other.” The mean baseline height standard deviation score (SDS) of -2.99 (1.02) in SOGROYA group and -3.47 (1.52) in daily somatropin group.
Treatment with once-weekly SOGROYA for 52 weeks resulted in an annualized height velocity of 11.2 cm/year. Patients treated with daily somatropin achieved an annualized height velocity of 11.7 cm/year after 52 weeks of treatment. Refer to Table 8.
Table 8. Annualized Height Velocity at Week 52 in Pediatric Patients with GHD
The mean increase in height SDS over the 52-week period was 1.25 and 1.30 in the once-weekly SOGROYA and daily somatropin groups, respectively.
14.2 Pediatric Patients Born Small for Gestational Age (SGA)
A multi-center, randomized, open-label, active-comparator, phase 3 basket study was conducted in GH treatment-naïve, pre-pubertal pediatric patients with short stature in small for gestational age (SGA), Noonan syndrome (NS) or idiopathic short stature (ISS) (NCT05330325). The primary efficacy endpoint was annualized height velocity at Week 52.
One hundred forty-two pediatric patients with SGA were randomized to SOGROYA 0.24 mg/kg/week (n=70), daily somatropin 0.035 mg/kg/day (n=37), or daily somatropin 0.067 mg/kg/day (n=35). Dose 0.035 mg/kg/day of daily somatropin is less than maximum dose (0.067 mg/kg/day) approved for use in pediatric patients with SGA in the United States. Patients ranged in age from 2.6 to 10.7 years with a mean of 5.5 years. Of these patients, 49% were male and 51% were female. Fifty-nine percent of patients were Caucasian, 36% of patients were Asian, 1% of patients were Black or African American, and 4% were not reported. The mean baseline height SDS was -3, -3, and -3 in the SOGROYA, somatropin 0.035 mg/kg/day, and somatropin 0.067 mg/kg/day groups, respectively.
The annualized height velocity at Week 52 for SOGROYA and somatropin are presented in Table 9.
Table 9. Annualized Height Velocity at Week 52 in Pediatric Patients with SGA
*Estimated treatment difference (Height Velocity of SOGROYA - Daily Somatropin)
#Height velocity at Week 52 is analyzed using an analysis of covariance model with treatment, gender, age group, region, baseline height SDS (<-3 or >=-3) and gender by age group by region interaction term as factors, and baseline height and baseline IGF-1 SDS as covariates. There were no missing values at Week 52, so no multiple imputation was done.
The mean increase in height SDS was 1.17, 0.85, and 1.21 following a 52-week treatment period with SOGROYA 0.24 mg/kg/week, daily somatropin 0.035 mg/kg/day, and daily somatropin 0.067 mg/kg/day, respectively.
14.3 Pediatric Patients with Noonan Syndrome (NS)
A multi-center, randomized, open-label, active-comparator, phase 3 basket study was conducted in GH treatment-naïve, pre-pubertal patients with short stature in small for gestational age (SGA), Noonan syndrome (NS) or idiopathic short stature (ISS) (NCT05330325). The primary efficacy endpoint was annualized height velocity at Week 52.
Seventy-seven NS patients were randomized to 0.24 mg/kg/week once weekly somapacitan-beco (n=49) or once daily somatropin at dose levels of 0.05 mg/kg/day (n=28). Dose 0.05 mg/kg/day of daily somatropin is less than maximum dose (0.066 mg/kg/day) approved for use in pediatric patients with NS in the United States. Patients ranged in age from 2 to 11.1 years with a mean of 6.2 years. Of these patients, 61% were male and 39% were female. Fifty-one percent of patients were Caucasian, 31% of patients were Asian, 5% of patients were Black or African American, and 12% were not reported, and 1% were reported as “multiple.” The mean baseline height SDS was -2.7 and -2.6 in the SOGROYA and somatropin 0.05 mg/kg/day groups, respectively.
The annualized height velocity at Week 52 was similar for somapacitan-beco and somatropin (Table 10).
Table 10. Annualized Height Velocity at Week 52 in Pediatric Patients with NS
*Estimated treatment difference (Height Velocity of SOGROYA - Daily Somatropin)
#Height velocity at Week 52 is analyzed using an analysis of covariance model with treatment, gender, age group, region, baseline height SDS (<-3 or >=-3) and gender by age group by region interaction term as factors, and baseline height and baseline IGF-1 SDS as covariates. Missing values at Week 52 are imputed based on available landmark visit data using Multiple Imputations.
The mean increase in height SDS was 1.06 and 0.79 following a 52-week treatment period with SOGROYA 0.24 mg/kg/week and daily somatropin 0.050 mg/kg/day, respectively.
14.4 Pediatric Patients with Idiopathic Short Stature (ISS)
A multi-center randomized, open-label, active-comparator, phase 3 basket study was conducted in GH treatment-naïve, pre-pubertal patients with short stature in small for gestational age (SGA), Noonan syndrome (NS) or idiopathic short stature (ISS) (NCT05330325). The primary efficacy endpoint was annualized height velocity at Week 52.
Eighty-eight ISS patients were randomized to 0.24 mg/kg/week once weekly somapacitan-beco (n=60) or once daily somatropin at dose levels of 0.05 mg/kg/day (n=28). Dose 0.05 mg/kg/day of daily somatropin is less than maximum dose (0.067 mg/kg/day) approved for use in pediatric patients with ISS in the United States. Patients ranged in age from 2.8 to 10.8 years with a mean of 6.9 years. Of these patients, 41% were male and 59% were female. Sixty-four percent (%) of patients were Caucasian, 30% of patients were Asian, 1% of patients were Black or African American, 1% were American Indian or Alaskan Native, 2% were not reported, and 2% were reported as “multiple.” The mean baseline height SDS was -2.8 and -2.9 in the SOGROYA and somatropin 0.05 mg/kg/day groups, respectively.
The annualized height velocity at Week 52 was similar for somapacitan-beco and somatropin (Table 11).
Table 11. Annualized Height Velocity at Week 52 in Pediatric Patients with ISS
*Estimated treatment difference (Height Velocity of SOGROYA - Daily Somatropin)
#Height velocity at Week 52 is analyzed using an analysis of covariance model with treatment, gender, age group, region, baseline height SDS (<-3 or >=-3) and gender by age group by region interaction term as factors, and baseline height and baseline IGF-1 SDS as covariates. Missing values at Week 52 are imputed based on available landmark visit data using Multiple Imputations.
The mean increase in height SDS was 0.98 and 1.09 following a 52-week treatment period with SOGROYA 0.24 mg/kg/week and daily somatropin 0.050 mg/kg/day, respectively.
14.5 Adults with Growth Hormone Deficiency (GHD)
In a 35-week, double-blind, placebo-controlled study, treatment-naïve adult patients with GHD were randomized (2:1:2) and exposed to once-weekly SOGROYA 10 mg/1.5mL (n=120) or placebo (n=60) or a daily somatropin product 10 mg/1.5mL (n=119) for a 34-week treatment period (NCT02229851).
In this study, patients were 51.7% female and had a mean age of 45.1 years. Most patients were 23 to 64 years old and most (69.7%) had adult onset GHD. The mean BMI was 27.4 kg/m2. Overall, 66.7% were White, 28.7% were Asian and 2.3% were Black or African American; 4.5% identified as Hispanic or Latino ethnicity.
Treatment with SOGROYA demonstrated superiority compared to placebo in reduction in truncal fat percentage (%) as assessed by dual X-ray absorptiometry, with a change of -1.06% for SOGROYA and +0.47% for placebo after 34 weeks (see Table 12). Patients treated with daily somatropin achieved a change in truncal fat % of -2.23% after 34 weeks.
Table 12. Truncal Fat % Results for Weekly SOGROYA, Weekly Placebo and Daily Somatropin During the 34-week Pivotal Trial
-
-
After 34 weeks SOGROYA normalized the mean IGF-1 SDS level in treatment-naïve adult patients with GHD with a IGF-1 SDS of -0.17 in SOGROYA-treated patients compared to -2.62 in placebo-treated patients (see Table 13). The mean IGF-1 SDS levels in daily somatropin-treated patients was -2.53 at baseline and -0.23 at 34 weeks.
Table 13. IGF-1 SDS for SOGROYA Compared to Placebo During the 34-week Pivotal Trial
Abbreviations: IGF-1 SDS: Insulin-like growth factor-1 standard deviation score, FAS = full analysis set, N = Number of patients in FAS. Baseline and end of main period (week 34) are observed means. Changes from baseline to the 35-week’s measurements were analysed using a mixed-effect model for repeated measurements including treatment, GHD onset type, sex, region, diabetes mellitus and sex by region by diabetes mellitus interaction as factors and baseline as a covariate, all nested within week as a factor.
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
SOGROYA (somapacitan-beco) injection is a clear to slightly opalescent and colorless to slightly yellow solution available as one 1.5 mL single-patient-use prefilled pen per carton:
-
SOGROYA 5 mg/1.5 mL (3.3 mg/mL) pen (teal) NDC 0169-2035-11 -
SOGROYA 10 mg/1.5 mL (6.7 mg/mL) pen (yellow) NDC 0169-2030-11 -
SOGROYA 15 mg/1.5 mL (10 mg/mL) pen (red) NDC 0169-2037-11 SOGROYA 5 mg/1.5 mL, 10 mg/1.5 mL, and 15 mg/1.5 mL pens are compatible with FlexPro® PenMate®. The FlexPro PenMate is an accessory device that is dispensed separately with its enclosed Instructions for Use.
Storage and Handling
Before and during use: Store in a refrigerator at 2°C to 8°C (36°F to 46°F) with the cap on and in the original carton to protect from light. Do not freeze. Do not use SOGROYA if it has been frozen. Discard prefilled pen if kept above 30°C (86°F). Avoid direct or excessive heat. Avoid sunlight. Refer to storage conditions for SOGROYA (Table 14).
Write the date of first use in the space provided on the carton.
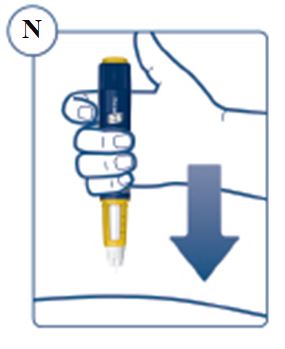
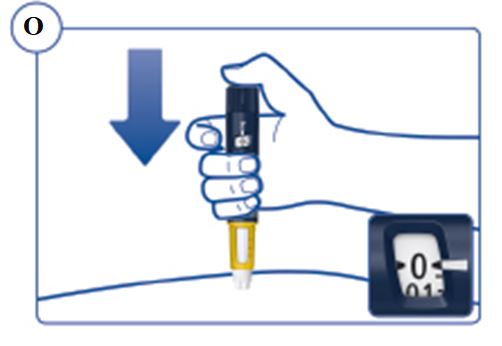
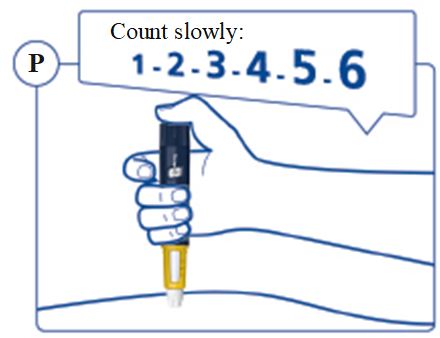
Always remove and safely discard the needle after each injection and store the SOGROYA prefilled pen without an injection needle attached. Always use a new needle for each injection to prevent contamination.
Table 14. Storage Conditions for SOGROYA
-
17 PATIENT COUNSELING INFORMATION
Advise patients and/or caregivers to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
-
Advise patients and/or caregivers to administer SOGROYA once weekly. -
Hypersensitivity - Advise patients and/or caregivers that severe and/or serious systemic hypersensitivity reactions (anaphylaxis and angioedema) have been reported, and to seek prompt medical attention should an allergic reaction occur [see Warnings and Precautions (5.2)]. -
Neoplasms – Advise patients to report marked changes in skin pigmentation or changes in the appearance of preexisting nevi. -
Glucose Intolerance/ Diabetes Mellitus – Advise patients that new onset pre- /diabetes mellitus or exacerbation of preexisting diabetes mellitus can occur and monitoring of blood glucose during treatment with SOGROYA may be needed. -
Intracranial Hypertension - Advise patients to report to their healthcare provider any visual changes, headache, and nausea and/or vomiting. -
Fluid Retention - Advise patients that fluid retention during SOGROYA replacement therapy may frequently occur. Inform patients of the clinical manifestations of fluid retention (e.g. edema, arthralgia, myalgia, nerve compression syndromes including carpal tunnel syndrome/paresthesia) and to report to their healthcare provider any of these signs or symptoms occur during treatment with SOGROYA. -
Hypoadrenalism - Advise patients who have or who are at risk for corticotropin deficiency that hypoadrenalism may develop and to report to their healthcare provider if they experience hyperpigmentation, extreme fatigue, dizziness, weakness, or weight loss. -
Hypothyroidism - Advise patients/caregivers that undiagnosed/untreated hypothyroidism may prevent an optimal response to SOGROYA. Advise patients/caregivers they may require periodic thyroid function tests. -
Pancreatitis - Advise patients that pancreatitis may develop and to report to their healthcare provider any new onset abdominal pain. -
Lipohypertrophy/ Lipoatrophy – Advise patients that lipohypertrophy or lipoatrophy can occur if SOGROYA is administered subcutaneously at the same site over a long period of time. Advise patients to rotate injection sites when administering SOGROYA to reduce this risk.
Novo Nordisk® and PenMate® are registered trademarks of Novo Nordisk A/S.
SOGROYA ® and FlexPro® are registered trademarks of Novo Nordisk Health Care AG.
© 2002-2026 Novo Nordisk Health Care AG
Patent Information: http://novonordisk-us.com/products/product-patents.html
For information about SOGROYA contact:
Novo Nordisk Inc.
800 Scudders Mill Road
Plainsboro, New Jersey 08536
1-888-668-6444
Manufactured by:
Novo Nordisk Inc.
800 Scudders Mill Road
Plainsboro, NJ 08536
U.S. License No. 1261
PATIENT INFORMATION
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 02/2026
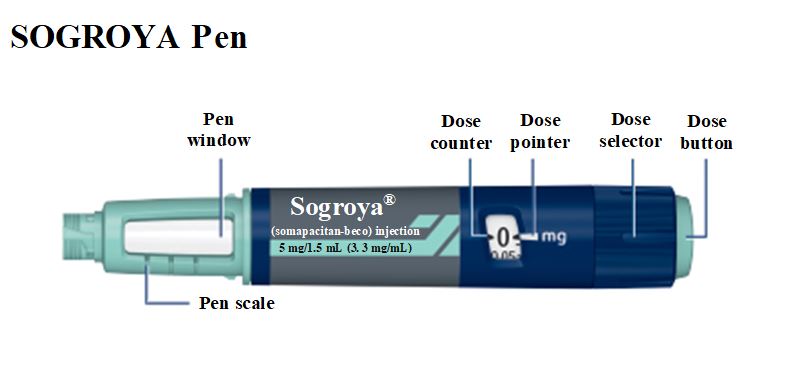
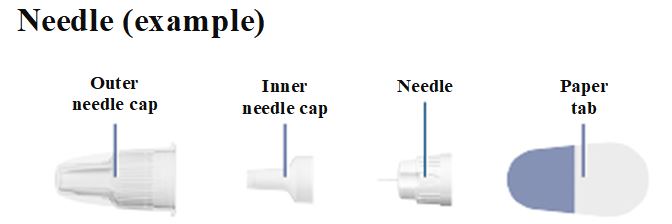
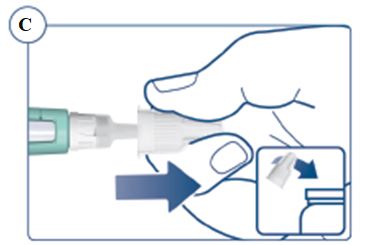
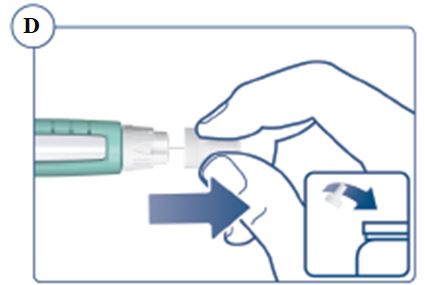
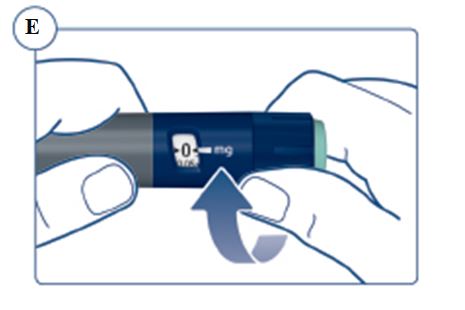
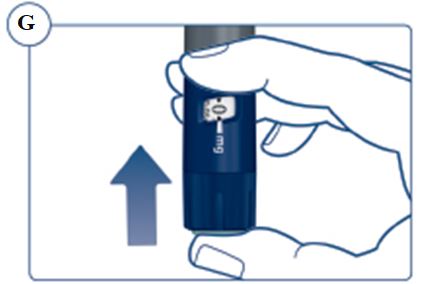
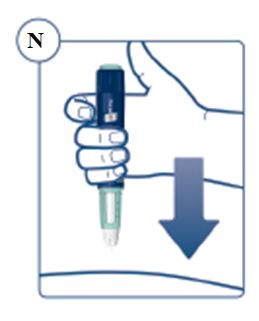
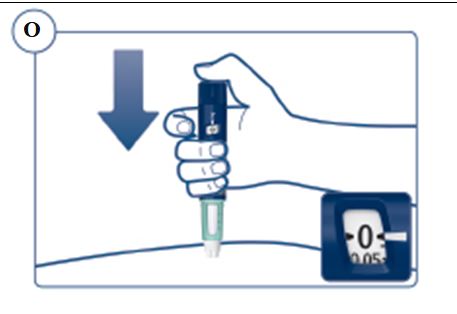
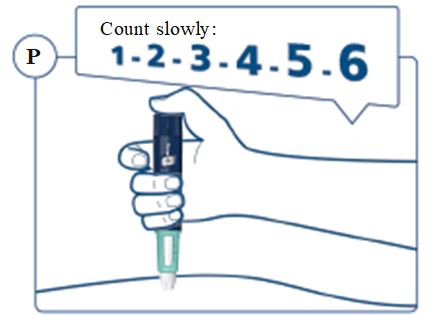
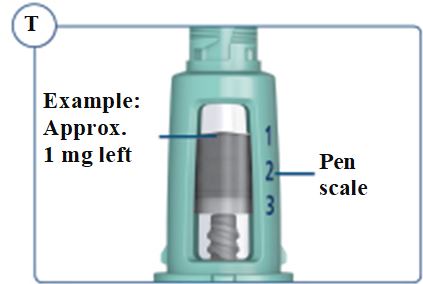
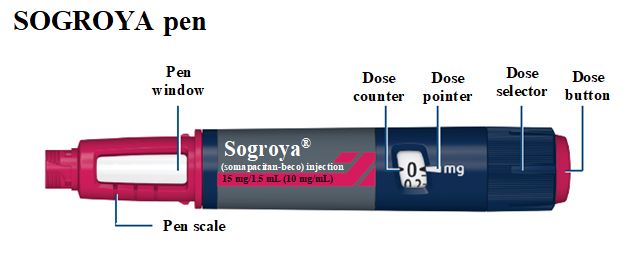
Instructions for Use – 5 mg/1.5 mL (3.3 mg/mL)
























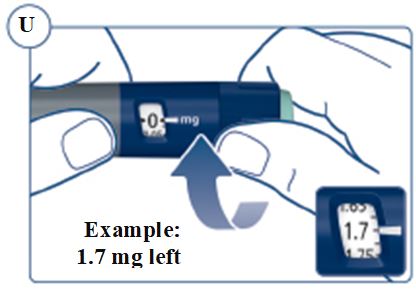

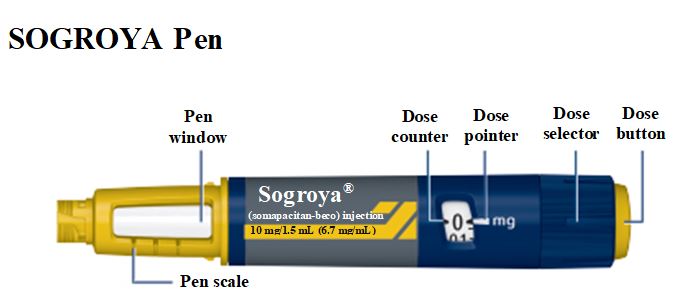
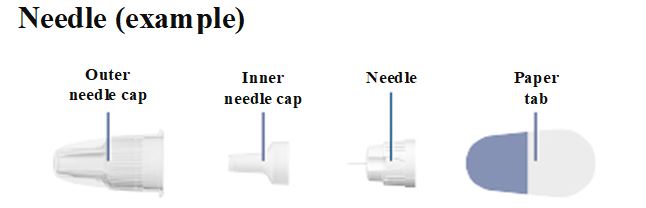
Instructions for Use – 10 mg/1.5 mL (6.7 mg/mL)



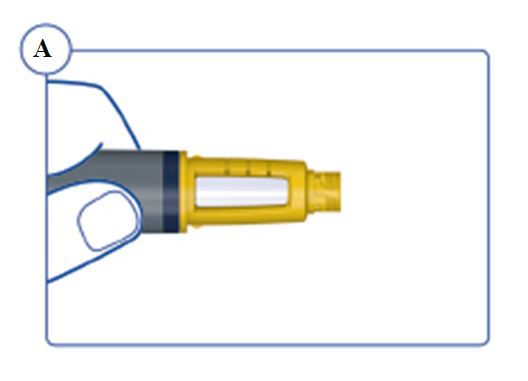
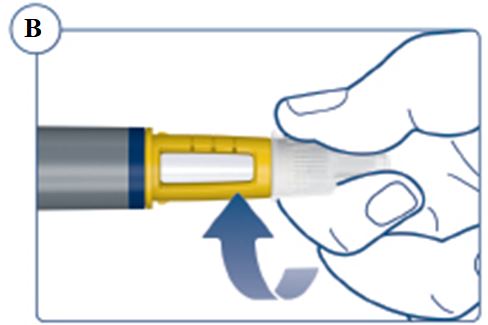


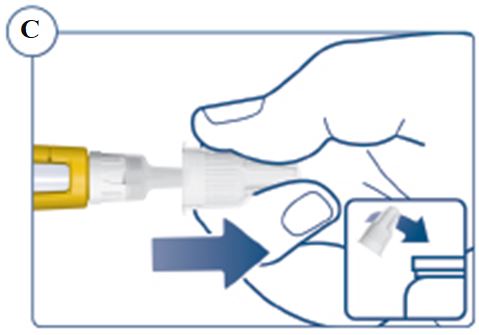

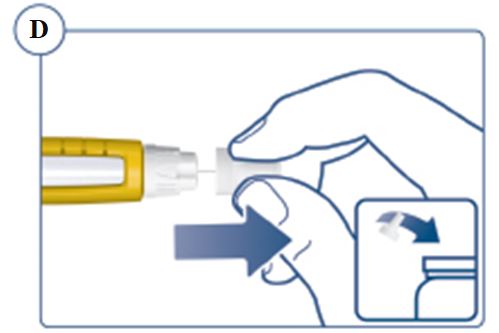


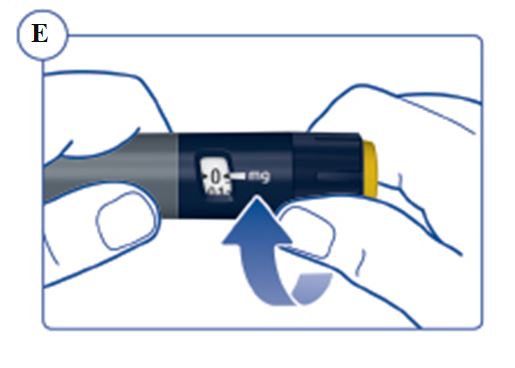

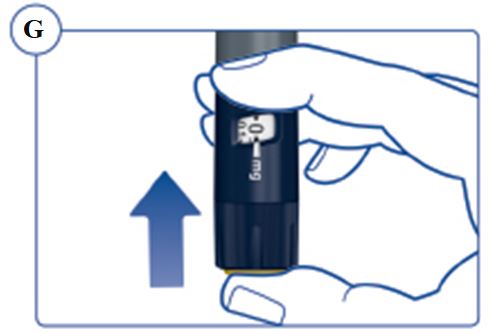



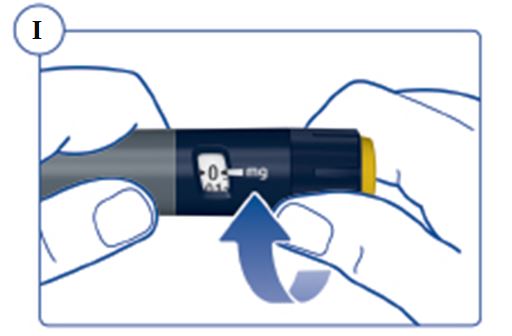




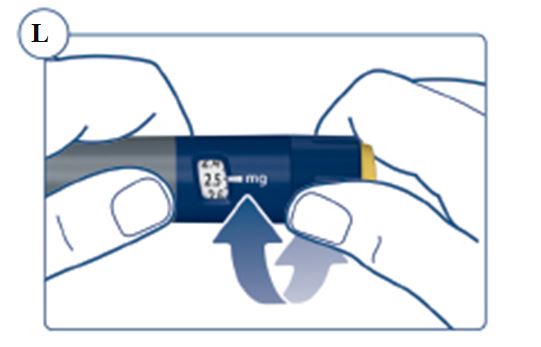


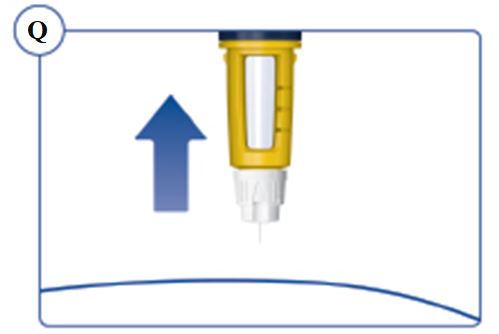

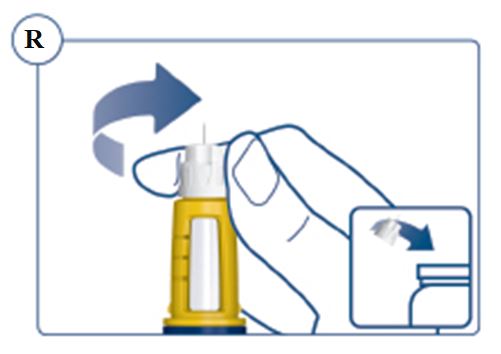
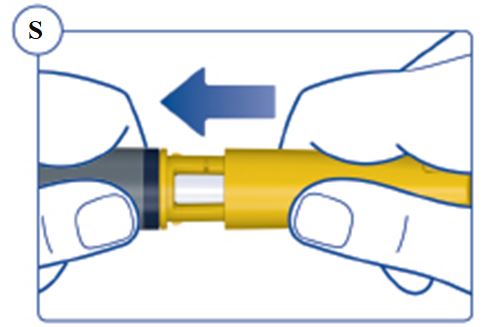


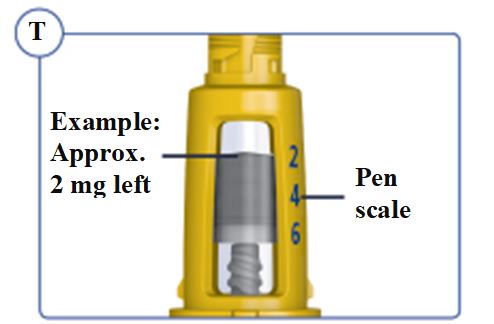
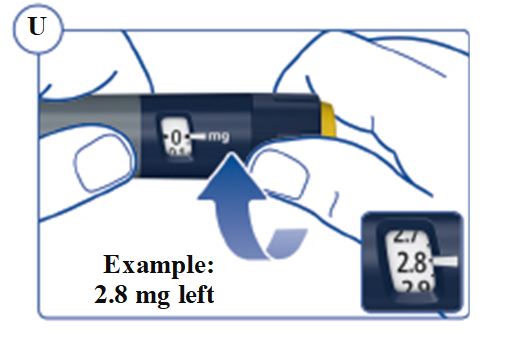

Instructions for Use – 15 mg/1.5 mL (10 mg/mL)




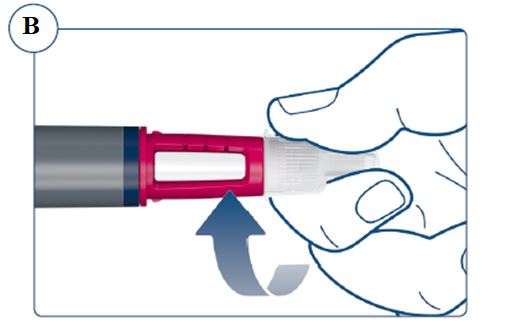


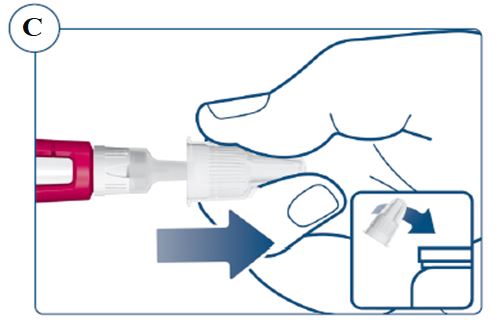

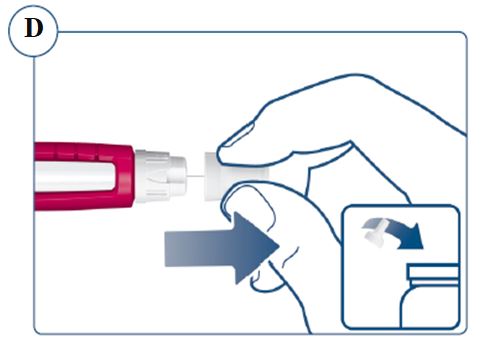




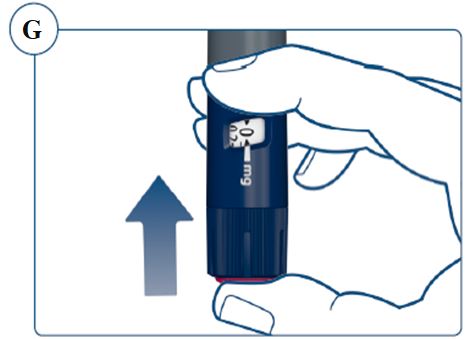



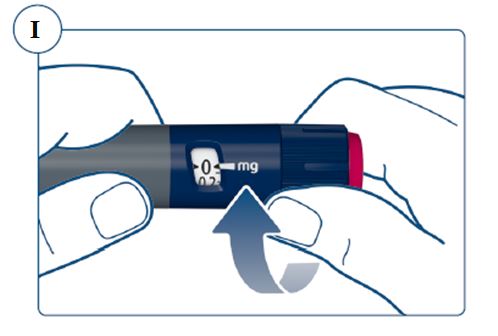




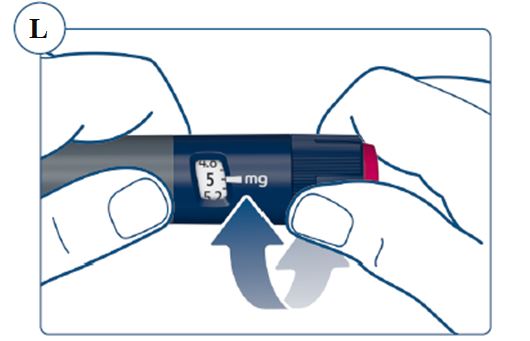
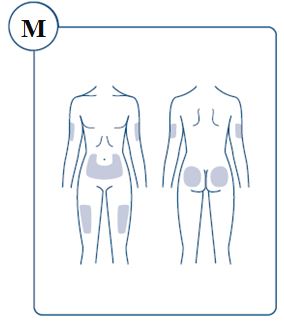
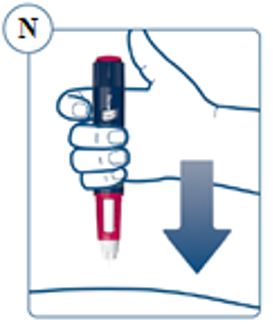

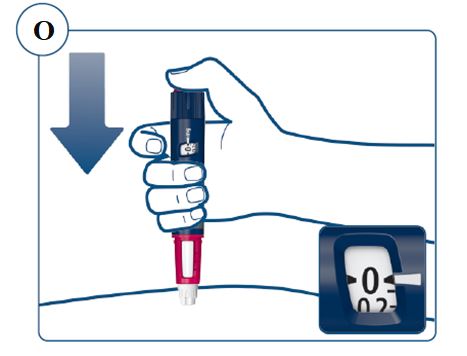
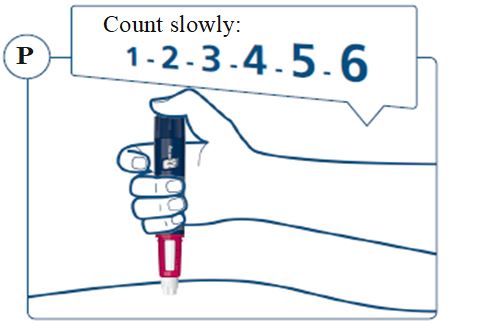

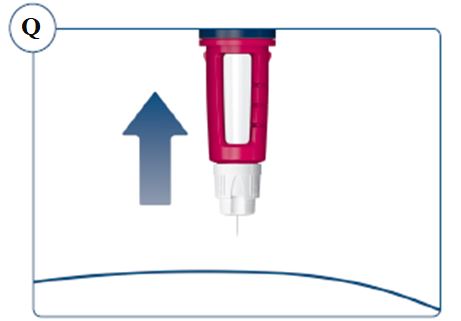

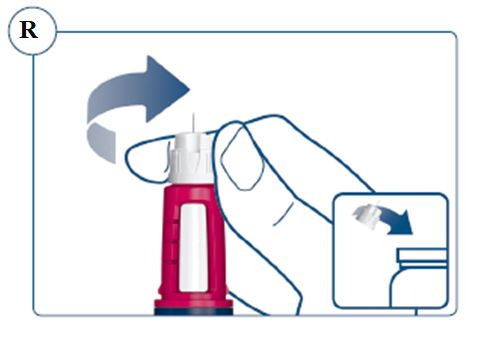
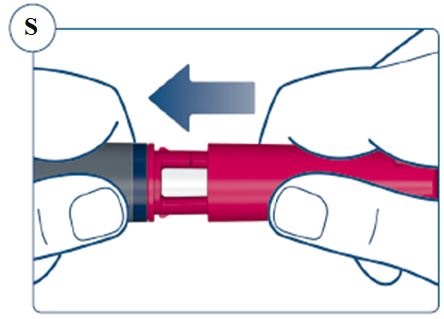



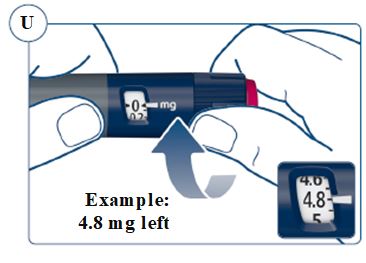


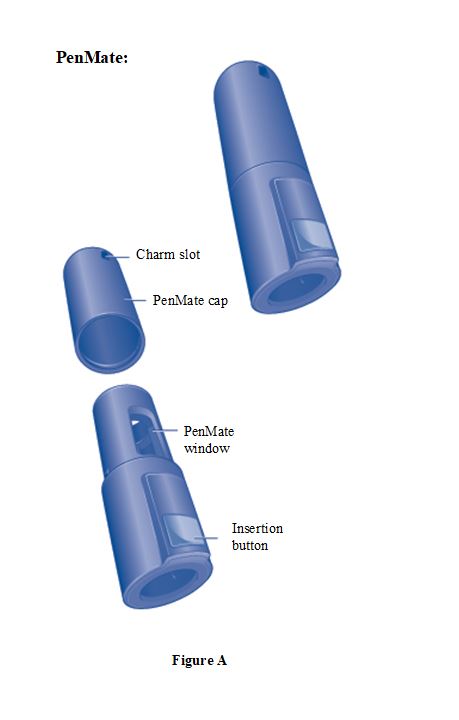
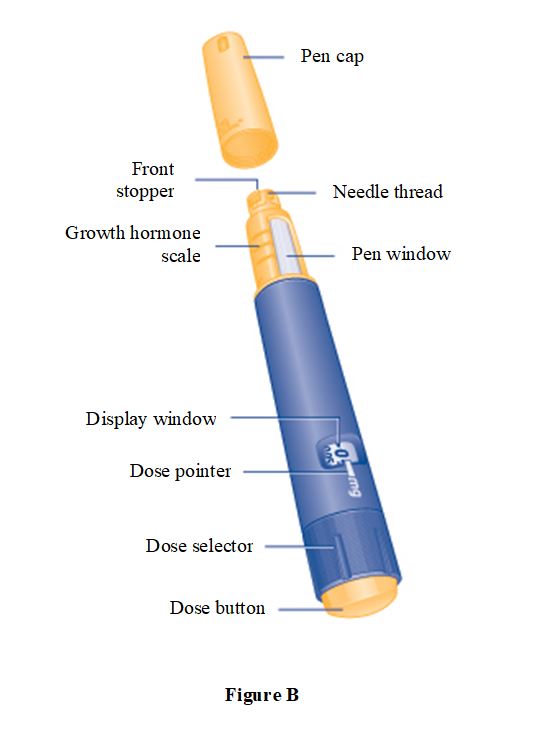
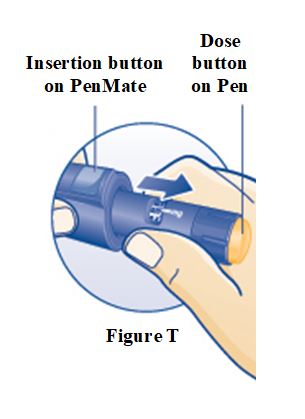
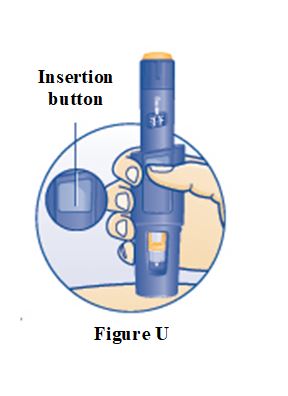
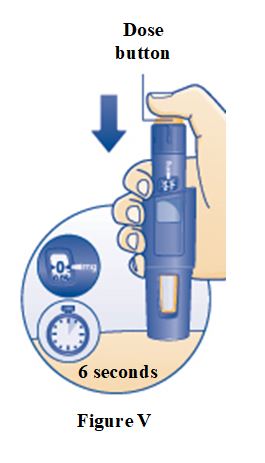
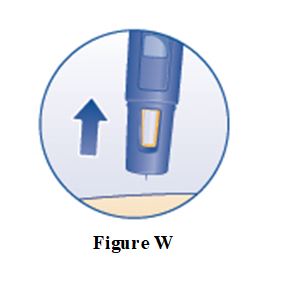
Instructions for Use – Norditropin® and Sogroya® with PenMate®
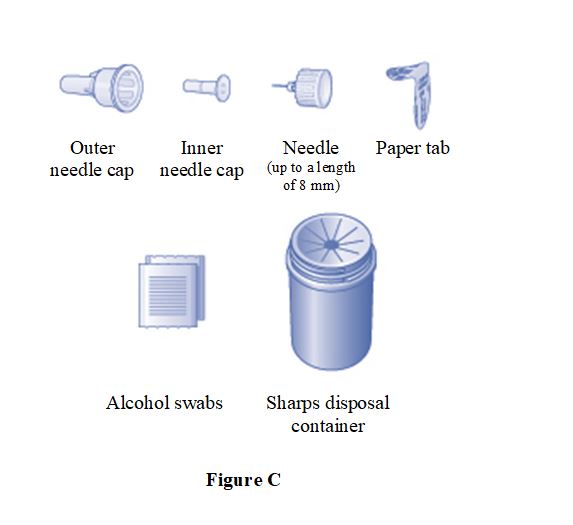

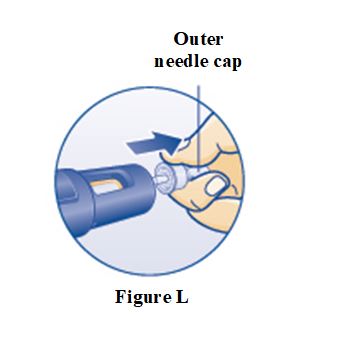
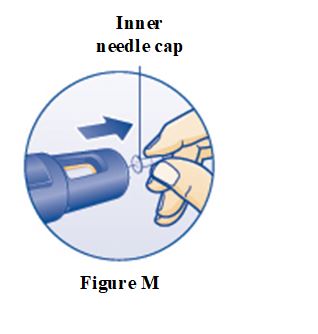
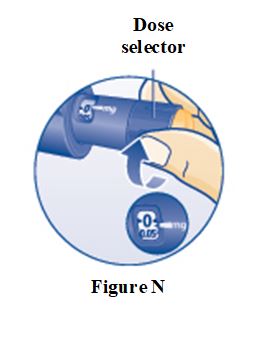
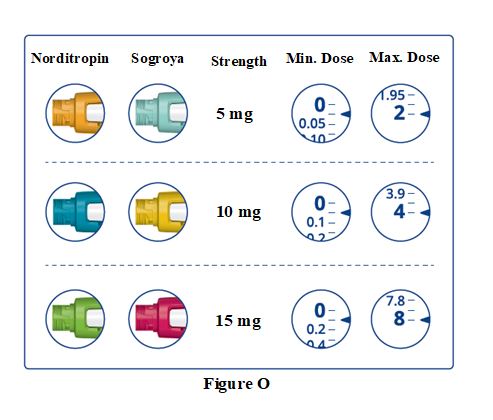
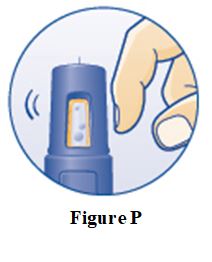
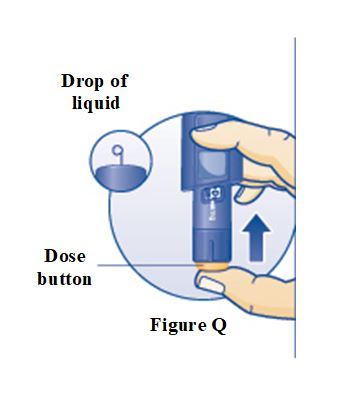
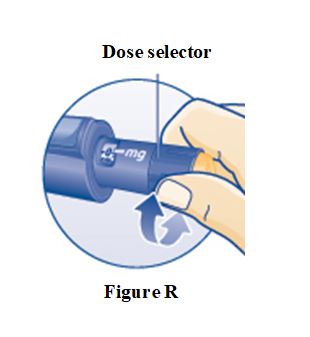

PRINCIPAL DISPLAY PANEL – 5 mg
-
-
5 mg / 1.5 mL (3.3 mg/mL) Prefilled Pen
For subcutaneous Use Only
1x1.5 mL single-patient use prefilled pen
Dials in 0.025 mg increments and contains 5 mg total
Rx only
once weekly

PRINCIPAL DISPLAY PANEL – 10 mg
-
-
10 mg / 1.5 mL (6.7 mg/mL) Prefilled Pen
For subcutaneous Use Only
1x1.5 mL single-patient use prefilled pen
Dials in 0.05 mg increments and contains 10 mg total
Rx only
once weekly

PRINCIPAL DISPLAY PANEL – 15 mg
-
-
15 mg / 1.5 mL (10 mg/mL) Prefilled Pen
For subcutaneous Use Only
1x1.5 mL single-patient use prefilled pen
Dials in 0.1 mg increments and contains 15 mg total
Rx only
once weekly

Guideline Central and select third party use “cookies” on this website to enhance the user experience.
This technology helps us gather statistical and analytical information to optimize the relevant content for you.
The user also has the option to opt-out which may have an effect on the browsing experience.