Alrex (loteprednol etabonate) suspension/ drops
Bausch & Lomb Incorporated
DESCRIPTION
ALREX® (loteprednol etabonate ophthalmic suspension, 0.2%) contains a sterile, topical anti-inflammatory corticosteroid for ophthalmic use. Loteprednol etabonate is a white to off-white powder.
Loteprednol etabonate is represented by the following structural formula:
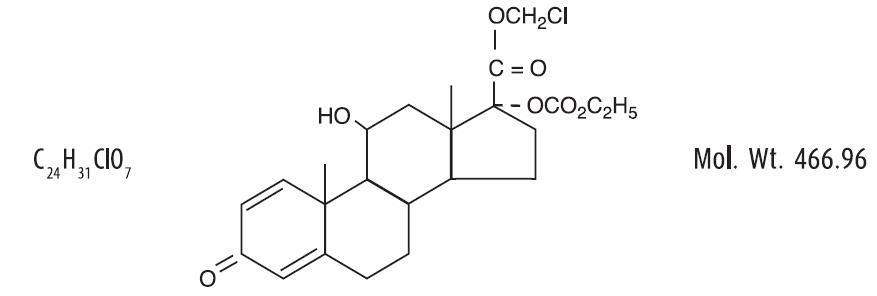
Chemical Name:
chloromethyl 17α-[(ethoxycarbonyl)oxy]-11β-hydroxy-3-oxoandrosta-1,4-diene-17β-carboxylate
Each mL contains:
ACTIVE: Loteprednol Etabonate 2 mg (0.2%);
INACTIVES: Edetate Disodium, Glycerin, Povidone, Purified Water and Tyloxapol. Hydrochloric Acid and/or Sodium Hydroxide may be added to adjust the pH. The suspension is essentially isotonic with a tonicity of 250 to 310 mOsmol/kg.
PRESERVATIVE ADDED: Benzalkonium Chloride 0.01%.
CLINICAL PHARMACOLOGY
Corticosteroids inhibit the inflammatory response to a variety of inciting agents and probably delay or slow healing. They inhibit the edema, fibrin deposition, capillary dilation, leukocyte migration, capillary proliferation, fibroblast proliferation, deposition of collagen, and scar formation associated with inflammation. There is no generally accepted explanation for the mechanism of action of ocular corticosteroids. However, corticosteroids are thought to act by the induction of phospholipase A2 inhibitory proteins, collectively called lipocortins. It is postulated that these proteins control the biosynthesis of potent mediators of inflammation such as prostaglandins and leukotrienes by inhibiting the release of their common precursor arachidonic acid. Arachidonic acid is released from membrane phospholipids by phospholipase A2. Corticosteroids are capable of producing a rise in intraocular pressure.
Loteprednol etabonate is structurally similar to other corticosteroids. However, the number 20 position ketone group is absent. It is highly lipid soluble which enhances its penetration into cells. Loteprednol etabonate is synthesized through structural modifications of prednisolone-related compounds so that it will undergo a predictable transformation to an inactive metabolite. Based upon in vivo and in vitro preclinical metabolism studies, loteprednol etabonate undergoes extensive metabolism to inactive carboxylic acid metabolites.
Results from a bioavailability study in normal volunteers established that plasma levels of loteprednol etabonate and Δ1 cortienic acid etabonate (PJ 91), its primary, inactive metabolite, were below the limit of quantitation (1 ng/mL) at all sampling times. The results were obtained following the ocular administration of one drop in each eye of 0.5% loteprednol etabonate 8 times daily for 2 days or 4 times daily for 42 days. This study suggests that limited (<1 ng/mL) systemic absorption occurs with ALREX.
Clinical Studies:
In two double-masked, placebo-controlled six-week environmental studies of 268 patients with seasonal allergic conjunctivitis, ALREX, when dosed four times per day was superior to placebo in the treatment of the signs and symptoms of seasonal allergic conjunctivitis. ALREX provided reduction in bulbar conjunctival injection and itching, beginning approximately 2 hours after instillation of the first dose and throughout the first 14 days of treatment.
INDICATIONS AND USAGE
ALREX ophthalmic suspension is indicated for the temporary relief of the signs and symptoms of seasonal allergic conjunctivitis.
CONTRAINDICATIONS
ALREX, as with other ophthalmic corticosteroids, is contraindicated in most viral diseases of the cornea and conjunctiva including epithelial herpes simplex keratitis (dendritic keratitis), vaccinia, and varicella, and also in mycobacterial infection of the eye and fungal diseases of ocular structures. ALREX is also contraindicated in individuals with known or suspected hypersensitivity to any of the ingredients of this preparation and to other corticosteroids.
WARNINGS
Prolonged use of corticosteroids may result in glaucoma with damage to the optic nerve, defects in visual acuity and fields of vision, and in posterior subcapsular cataract formation. Steroids should be used with caution in the presence of glaucoma.
Prolonged use of corticosteroids may suppress the host response and thus increase the hazard of secondary ocular infections. In those diseases causing thinning of the cornea or sclera, perforations have been known to occur with the use of topical steroids. In acute purulent conditions of the eye, steroids may mask infection or enhance existing infection.
Use of ocular steroids may prolong the course and may exacerbate the severity of many viral infections of the eye (including herpes simplex). Employment of a corticosteroid medication in the treatment of patients with a history of herpes simplex requires great caution.
PRECAUTIONS
General:
For ophthalmic use only. The initial prescription and renewal of the medication order beyond 14 days should be made by a physician only after examination of the patient with the aid of magnification, such as slit lamp biomicroscopy and, where appropriate, fluorescein staining.
If signs and symptoms fail to improve after two days, the patient should be re-evaluated.
If this product is used for 10 days or longer, intraocular pressure should be monitored.
Fungal infections of the cornea are particularly prone to develop coincidentally with long-term local steroid application. Fungus invasion must be considered in any persistent corneal ulceration where a steroid has been used or is in use. Fungal cultures should be taken when appropriate.
Information for Patients:
This product is sterile when packaged. Patients should be advised not to allow the dropper tip to touch any surface, as this may contaminate the suspension. If redness or itching becomes aggravated, the patient should be advised to consult a physician.
Patients should be advised not to wear a contact lens if their eye is red. ALREX should not be used to treat contact lens related irritation. The preservative in ALREX, benzalkonium chloride, may be absorbed by soft contact lenses. Patients who wear soft contact lenses and whose eyes are not red, should be instructed to wait at least ten minutes after instilling ALREX before they insert their contact lenses.
Carcinogenesis, Mutagenesis, Impairment of Fertility:
Long-term animal studies have not been conducted to evaluate the carcinogenic potential of loteprednol etabonate. Loteprednol etabonate was not genotoxic in vitro in the Ames test, the mouse lymphoma tk assay, or in a chromosome aberration test in human lymphocytes, or in vivo in the single dose mouse micronucleus assay. Treatment of male and female rats with up to 50 mg/kg/day and 25 mg/kg/day of loteprednol etabonate, respectively, (1,500 and 750 times the maximum clinical dose, respectively) prior to and during mating did not impair fertility in either gender.
Pregnancy:
Teratogenic effects:
Loteprednol etabonate has been shown to be embryotoxic (delayed ossification) and teratogenic (increased incidence of meningocele, abnormal left common carotid artery, and limb flexures) when administered orally to rabbits during organogenesis at a dose of 3 mg/kg/day (85 times the maximum daily clinical dose), a dose which caused no maternal toxicity. The no-observed-effect-level (NOEL) for these effects was 0.5 mg/kg/day (15 times the maximum daily clinical dose). Oral treatment of rats during organogenesis resulted in teratogenicity (absent innominate artery at ≥5 mg/kg/day doses, and cleft palate and umbilical hernia at ≥50 mg/kg/day) and embryotoxicity (increased postimplantation losses at 100 mg/kg/day and decreased fetal body weight and skeletal ossification with ≥50 mg/kg/day). Treatment of rats with 0.5 mg/kg/day (15 times the maximum clinical dose) during organogenesis did not result in any reproductive toxicity. Loteprednol etabonate was maternally toxic (significantly reduced body weight gain during treatment) when administered to pregnant rats during organogenesis at doses of ≥5 mg/kg/day.
Oral exposure of female rats to 50 mg/kg/day of loteprednol etabonate from the start of the fetal period through the end of lactation, a maternally toxic treatment regimen (significantly decreased body weight gain), gave rise to decreased growth and survival, and retarded development in the offspring during lactation; the NOEL for these effects was 5 mg/kg/day. Loteprednol etabonate had no effect on the duration of gestation or parturition when administered orally to pregnant rats at doses up to 50 mg/kg/day during the fetal period.
There are no adequate and well controlled studies in pregnant women. ALREX ophthalmic suspension should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers:
It is not known whether topical ophthalmic administration of corticosteroids could result in sufficient systemic absorption to produce detectable quantities in human milk. Systemic steroids appear in human milk and could suppress growth, interfere with endogenous corticosteroid production, or cause other untoward effects. Caution should be exercised when ALREX is administered to a nursing woman.
Pediatric Use:
Safety and effectiveness in pediatric patients have not been established.
ADVERSE REACTIONS
Reactions associated with ophthalmic steroids include elevated intraocular pressure, which may be associated with optic nerve damage, visual acuity and field defects, posterior subcapsular cataract formation, secondary ocular infection from pathogens including herpes simplex, and perforation of the globe where there is thinning of the cornea or sclera.
Ocular adverse reactions occurring in 5-15% of patients treated with loteprednol etabonate ophthalmic suspension (0.2-0.5%) in clinical studies included abnormal vision/blurring, burning on instillation, chemosis, discharge, dry eyes, epiphora, foreign body sensation, itching, injection, and photophobia. Other ocular adverse reactions occurring in less than 5% of patients include conjunctivitis, corneal abnormalities, eyelid erythema, keratoconjunctivitis, ocular irritation/pain/discomfort, papillae, and uveitis. Some of these events were similar to the underlying ocular disease being studied.
Non-ocular adverse reactions occurred in less than 15% of patients. These include headache, rhinitis and pharyngitis.
In a summation of controlled, randomized studies of individuals treated for 28 days or longer with loteprednol etabonate, the incidence of significant elevation of intraocular pressure (≥10 mm Hg) was 2% (15/901) among patients receiving loteprednol etabonate, 7% (11/164) among patients receiving 1% prednisolone acetate and 0.5% (3/583) among patients receiving placebo. Among the smaller group of patients who were studied with ALREX, the incidence of clinically significant increases in IOP (≥10 mm Hg) was 1% (1/133) with ALREX and 1% (1/135) with placebo.
To report SUSPECTED ADVERSE REACTIONS, contact Bausch & Lomb Incorporated at 1-800-553-5340 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DOSAGE AND ADMINISTRATION
SHAKE VIGOROUSLY BEFORE USING.
One drop instilled into the affected eye(s) four times daily.
HOW SUPPLIED
ALREX® (loteprednol etabonate ophthalmic suspension, 0.2%) is supplied in a plastic bottle with a controlled drop tip in the following sizes:
-
-
Storage:
Store upright between 15°C to 25°C (59°F to 77°F). DO NOT FREEZE.

Distributed by:
Bausch & Lomb Americas Inc.
Bridgewater, NJ 08807 USA
Manufactured by:
Bausch & Lomb Incorporated
Tampa, FL 33637 USA
ALREX is a trademark of Bausch & Lomb Incorporated or its affiliates.
© 2022 Bausch & Lomb Incorporated or its affiliates
Revised: 03/2022
9007907 (Folded)
9005507 (Flat)
Principal Display Panel

9537903
AB35309
NDC 24208-353-10
Alrex
®
loteprednol etabonate
ophthalmic suspension, 0.2%
FOR OPHTHALMIC USE ONLY
Sterile
Rx only
10 mL