Procardia (nifedipine) capsule
Pfizer Laboratories Div Pfizer Inc
DESCRIPTION
PROCARDIA® (nifedipine) is an antianginal drug belonging to a class of pharmacological agents, the calcium channel blockers. Nifedipine is 3,5-pyridinedicarboxylic acid, 1,4-dihydro-2,6-dimethyl-4-(2-nitrophenyl)-, dimethyl ester, C17H18N2O6, and has the structural formula:
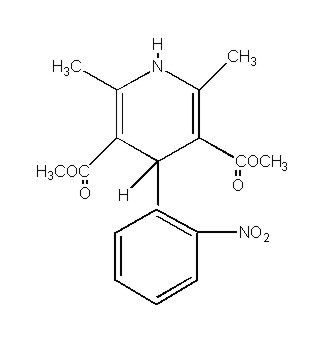
Nifedipine is a yellow crystalline substance, practically insoluble in water but soluble in ethanol. It has a molecular weight of 346.3. PROCARDIA capsules are formulated as soft gelatin capsules for oral administration, each containing 10 mg nifedipine.
Inert ingredients in the formulation are glycerin; peppermint oil; polyethylene glycol; soft gelatin capsules (which contain Yellow 6, and may contain Red Ferric Oxide and other inert ingredients); and water. The 10 mg capsules also contain saccharin sodium.
CLINICAL PHARMACOLOGY
PROCARDIA is a calcium ion influx inhibitor (slow-channel blocker or calcium ion antagonist) and inhibits the transmembrane influx of calcium ions into cardiac muscle and smooth muscle. The contractile processes of cardiac muscle and vascular smooth muscle are dependent upon the movement of extracellular calcium ions into these cells through specific ion channels. PROCARDIA selectively inhibits calcium ion influx across the cell membrane of cardiac muscle and vascular smooth muscle without changing serum calcium concentrations.
Mechanism of Action
The precise means by which this inhibition relieves angina has not been fully determined, but includes at least the following two mechanisms:
1) Relaxation and Prevention of Coronary Artery Spasm
PROCARDIA dilates the main coronary arteries and coronary arterioles, both in normal and ischemic regions, and is a potent inhibitor of coronary artery spasm, whether spontaneous or ergonovine-induced. This property increases myocardial oxygen delivery in patients with coronary artery spasm, and is responsible for the effectiveness of PROCARDIA in vasospastic (Prinzmetal's or variant) angina. Whether this effect plays any role in classical angina is not clear, but studies of exercise tolerance have not shown an increase in the maximum exercise rate-pressure product, a widely accepted measure of oxygen utilization. This suggests that, in general, relief of spasm or dilation of coronary arteries is not an important factor in classical angina.
2) Reduction of Oxygen Utilization
PROCARDIA regularly reduces arterial pressure at rest and at a given level of exercise by dilating peripheral arterioles and reducing the total peripheral resistance (afterload) against which the heart works. This unloading of the heart reduces myocardial energy consumption and oxygen requirements and probably accounts for the effectiveness of PROCARDIA in chronic stable angina.
Pharmacokinetics and Metabolism
PROCARDIA is rapidly and fully absorbed after oral administration. The drug is detectable in serum 10 minutes after oral administration, and peak blood levels occur in approximately 30 minutes. Bioavailability is proportional to dose from 10 to 30 mg; half-life does not change significantly with dose. There is little difference in relative bioavailability when PROCARDIA capsules are given orally and either swallowed whole, bitten and swallowed, or bitten and held sublingually. However, biting through the capsule prior to swallowing does result in slightly earlier plasma concentrations (27 ng/mL 10 minutes after 10 mg) than if capsules are swallowed intact. PROCARDIA is highly bound by serum proteins. PROCARDIA is extensively converted to inactive metabolites and approximately 80 percent of PROCARDIA and metabolites are eliminated via the kidneys. The elimination half-life of nifedipine is approximately two hours. Since hepatic biotransformation is the predominant route for the disposition of nifedipine, the pharmacokinetics may be altered in patients with chronic liver disease. Patients with hepatic impairment (liver cirrhosis) have a longer disposition half-life and higher bioavailability of nifedipine than healthy volunteers. The degree of serum protein binding of nifedipine is high (92–98%). Protein binding may be greatly reduced in patients with renal or hepatic impairment.
Following intravenous administration, clearance of nifedipine was decreased by 33% in elderly healthy subjects relative to young healthy subjects.
Hemodynamics
Like other slow-channel blockers, PROCARDIA exerts a negative inotropic effect on isolated myocardial tissue. This is rarely, if ever, seen in intact animals or man, probably because of reflex responses to its vasodilating effects. In man, PROCARDIA causes decreased peripheral vascular resistance and a fall in systolic and diastolic pressure, usually modest (5–10 mm Hg systolic), but sometimes larger. There is usually a small increase in heart rate, a reflex response to vasodilation. Measurements of cardiac function in patients with normal ventricular function have generally found a small increase in cardiac index without major effects on ejection fraction, left ventricular end diastolic pressure (LVEDP), or volume (LVEDV). In patients with impaired ventricular function, most acute studies have shown some increase in ejection fraction and reduction in left ventricular filling pressure.
Electrophysiologic Effects
Although, like other members of its class, PROCARDIA decreases sinoatrial node function and atrioventricular conduction in isolated myocardial preparations, such effects have not been seen in studies in intact animals or in man. In formal electrophysiologic studies, predominantly in patients with normal conduction systems, PROCARDIA has had no tendency to prolong atrioventricular conduction, prolong sinus node recovery time, or slow sinus rate.
INDICATIONS AND USAGE
I. Vasospastic Angina
PROCARDIA (nifedipine) is indicated for the management of vasospastic angina confirmed by any of the following criteria: 1) classical pattern of angina at rest accompanied by ST segment elevation, 2) angina or coronary artery spasm provoked by ergonovine, or 3) angiographically demonstrated coronary artery spasm. In those patients who have had angiography, the presence of significant fixed obstructive disease is not incompatible with the diagnosis of vasospastic angina, provided that the above criteria are satisfied. PROCARDIA may also be used where the clinical presentation suggests a possible vasospastic component but where vasospasm has not been confirmed, e.g., where pain has a variable threshold on exertion or when angina is refractory to nitrates and/or adequate doses of beta blockers.
II. Chronic Stable Angina
(Classical Effort-Associated Angina)
PROCARDIA is indicated for the management of chronic stable angina (effort-associated angina) without evidence of vasospasm in patients who remain symptomatic despite adequate doses of beta blockers and/or organic nitrates or who cannot tolerate those agents.
In chronic stable angina (effort-associated angina), PROCARDIA has been effective in controlled trials of up to eight weeks duration in reducing angina frequency and increasing exercise tolerance, but confirmation of sustained effectiveness and evaluation of long-term safety in these patients are incomplete.
Controlled studies in small numbers of patients suggest concomitant use of PROCARDIA and beta-blocking agents may be beneficial in patients with chronic stable angina, but available information is not sufficient to predict with confidence the effects of concurrent treatment, especially in patients with compromised left ventricular function or cardiac conduction abnormalities. When introducing such concomitant therapy, care must be taken to monitor blood pressure closely since severe hypotension can occur from the combined effects of the drugs. (See WARNINGS.)
CONTRAINDICATIONS
Known hypersensitivity reaction to PROCARDIA.
WARNINGS
Excessive Hypotension
Although, in most patients, the hypotensive effect of PROCARDIA is modest and well tolerated, occasional patients have had excessive and poorly tolerated hypotension. These responses have usually occurred during initial titration or at the time of subsequent upward dosage adjustment. Although patients have rarely experienced excessive hypotension on PROCARDIA alone, this may be more common in patients on concomitant beta blocker therapy. Although not approved for this purpose, PROCARDIA and other immediate-release nifedipine capsules have been used (orally and sublingually) for acute reduction of blood pressure. Several well-documented reports describe cases of profound hypotension, myocardial infarction, and death when immediate-release nifedipine was used in this way. PROCARDIA capsules should not be used for the acute reduction of blood pressure.
Severe hypotension and/or increased fluid volume requirements have been reported in patients receiving PROCARDIA together with a beta-blocking agent who underwent coronary artery bypass surgery using high dose fentanyl anesthesia. The interaction with high dose fentanyl appears to be due to the combination of PROCARDIA and a beta blocker, but the possibility that it may occur with PROCARDIA alone, with low doses of fentanyl, in other surgical procedures, or with other narcotic analgesics cannot be ruled out. In PROCARDIA treated patients where surgery using high dose fentanyl anesthesia is contemplated, the physician should be aware of these potential problems and, if the patient's condition permits, sufficient time (at least 36 hours) should be allowed for PROCARDIA to be washed out of the body prior to surgery.
Increased Angina and/or Myocardial Infarction
Rarely, patients, particularly those who have severe obstructive coronary artery disease, have developed well documented increased frequency, duration, and/or severity of angina or acute myocardial infarction on starting PROCARDIA or at the time of dosage increase. The mechanism of this effect is not established.
Several well-controlled, randomized trials studied the use of immediate-release nifedipine in patients who had just sustained myocardial infarctions. In none of these trials did immediate-release nifedipine appear to provide any benefit. In some of the trials, patients who received immediate-release nifedipine had significantly worse outcomes than patients who received placebo. PROCARDIA capsules should not be administered within the first week or two after myocardial infarction, and they should also be avoided in the setting of acute coronary syndrome (when infarction may be imminent).
Use in Essential Hypertension
PROCARDIA and other immediate-release nifedipine capsules have also been used for the long-term control of essential hypertension, although PROCARDIA capsules have not been approved for this purpose and no properly controlled studies have been conducted to define an appropriate dose or dose interval for such treatment. PROCARDIA capsules should not be used for the control of essential hypertension.
Beta Blocker Withdrawal
Patients recently withdrawn from beta blockers may develop a withdrawal syndrome with increased angina, probably related to increased sensitivity to catecholamines. Initiation of PROCARDIA treatment will not prevent this occurrence and might be expected to exacerbate it by provoking reflex catecholamine release. There have been occasional reports of increased angina in a setting of beta blocker withdrawal and PROCARDIA initiation. It is important to taper beta blockers if possible, rather than stopping them abruptly before beginning PROCARDIA.
Congestive Heart Failure
Rarely, patients, usually those receiving a beta blocker, have developed heart failure after beginning PROCARDIA. Patients with tight aortic stenosis may be at greater risk for such an event, as the unloading effect of PROCARDIA would be expected to be of less benefit to these patients, owing to their fixed impedance to flow across the aortic valve.
PRECAUTIONS
General
Hypotension
Because PROCARDIA decreases peripheral vascular resistance, careful monitoring of blood pressure during the initial administration and titration of PROCARDIA is suggested. Close observation is especially recommended for patients already taking medications that are known to lower blood pressure. (See WARNINGS.)
Peripheral Edema
Mild to moderate peripheral edema, typically associated with arterial vasodilation and not due to left ventricular dysfunction, occurs in about one in ten patients treated with PROCARDIA (nifedipine). This edema occurs primarily in the lower extremities and usually responds to diuretic therapy. With patients whose angina is complicated by congestive heart failure, care should be taken to differentiate this peripheral edema from the effects of increasing left ventricular dysfunction.
Laboratory Tests
Rare, usually transient, but occasionally significant elevations of enzymes such as alkaline phosphatase, CPK, LDH, SGOT, and SGPT have been noted. The relationship to PROCARDIA therapy is uncertain in most cases, but probable in some. These laboratory abnormalities have rarely been associated with clinical symptoms; however, cholestasis with or without jaundice has been reported. Rare instances of allergic hepatitis have been reported.
PROCARDIA, like other calcium channel blockers, decreases platelet aggregation in vitro. Limited clinical studies have demonstrated a moderate but statistically significant decrease in platelet aggregation and an increase in bleeding time in some PROCARDIA patients. This is thought to be a function of inhibition of calcium transport across the platelet membrane. No clinical significance for these findings has been demonstrated.
Positive direct Coombs Test with/without hemolytic anemia has been reported but a causal relationship between PROCARDIA administration and positivity of this laboratory test, including hemolysis, could not be determined.
Although PROCARDIA has been used safely in patients with renal dysfunction and has been reported to exert a beneficial effect, in certain cases, rare, reversible elevations in BUN and serum creatinine have been reported in patients with pre-existing chronic renal insufficiency. The relationship to PROCARDIA therapy is uncertain in most cases but probable in some.
Drug Interactions
Beta-adrenergic blocking agents
(See INDICATIONS AND USAGE and WARNINGS.) Experience in over 1400 patients in a non-comparative clinical trial has shown that concomitant administration of PROCARDIA and beta-blocking agents is usually well tolerated, but there have been occasional literature reports suggesting that the combination may increase the likelihood of congestive heart failure, severe hypotension, or exacerbation of angina.
Long-acting nitrates
PROCARDIA may be safely co-administered with nitrates, but there have been no controlled studies to evaluate the antianginal effectiveness of this combination.
Digitalis
Since there have been isolated reports of patients with elevated digoxin levels, and since there is a possible interaction between digoxin and nifedipine, it is recommended that digoxin levels be monitored when initiating, adjusting, and discontinuing nifedipine to avoid possible over- or under-digitalization.
Quinidine
There have been rare reports of an interaction between quinidine and nifedipine (with a decreased plasma level of quinidine).
Coumarin anticoagulants
There have been rare reports of increased prothrombin time in patients taking coumarin anticoagulants to whom PROCARDIA was administered. However, the relationship to PROCARDIA therapy is uncertain.
Cimetidine
A study in six healthy volunteers has shown a significant increase in peak nifedipine plasma levels (80%) and area-under-the-curve (74%) after a one week course of cimetidine at 1000 mg per day and nifedipine at 40 mg per day. Ranitidine produced smaller, non-significant increases. The effect may be mediated by the known inhibition of cimetidine on hepatic cytochrome P-450, the enzyme system probably responsible for the first-pass metabolism of nifedipine. If nifedipine therapy is initiated in a patient currently receiving cimetidine, cautious titration is advised.
Nifedipine is metabolized by CYP3A4. Co-administration of nifedipine with phenytoin, an inducer of CYP3A4, lowers the systemic exposure to nifedipine by approximately 70%. Avoid co-administration of nifedipine with phenytoin or any known CYP3A4 inducer or consider an alternative antihypertensive therapy.
CYP3A inhibitors such as fluconazole, itraconazole, clarithromycin, erythromycin, nefazodone, fluoxetine, saquinavir, indinavir, and nelfinavir may result in increased exposure to nifedipine when co-administered. Careful monitoring and dose adjustment may be necessary; consider initiating nifedipine at the lowest dose available if given concomitantly with these medications.
Other Interactions
Grapefruit Juice
Co-administration of nifedipine with grapefruit juice resulted in approximately a doubling in nifedipine AUC and Cmax with no change in half-life. The increased plasma concentrations most likely result from inhibition of CYP 3A4 related first-pass metabolism. Avoid ingestion of grapefruit and grapefruit juice while taking nifedipine.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Nifedipine was administered orally to rats for two years and was not shown to be carcinogenic. When given to rats prior to mating, nifedipine caused reduced fertility at a dose approximately 5 times the maximum recommended human dose. There is a literature report of reversible reduction in the ability of human sperm obtained from a limited number of infertile men taking recommended doses of nifedipine to bind to and fertilize an ovum in vitro. In vivo mutagenicity studies were negative.
Pregnancy
Nifedipine has been shown to produce teratogenic findings in rats and rabbits, including digital anomalies similar to those reported for phenytoin. Digital anomalies have been reported to occur with other members of the dihydropyridine class and are possibly a result of compromised uterine blood flow. Nifedipine administration was associated with a variety of embryotoxic, placentotoxic, and fetotoxic effects, including stunted fetuses (rats, mice, rabbits), rib deformities (mice), cleft palate (mice), small placentas and underdeveloped chorionic villi (monkeys), embryonic and fetal deaths (rats, mice, rabbits), and prolonged pregnancy/decreased neonatal survival (rats; not evaluated in other species). On a mg/kg basis, all of the doses associated with the teratogenic embryotoxic or fetotoxic effects in animals were higher (5 to 50 times) than the maximum recommended human dose of 120 mg/day. On a mg/m2 basis, some doses were higher and some were lower than the maximum recommended human dose but all were within an order of magnitude of it. The doses associated with placentotoxic effects in monkeys were equivalent to or lower than the maximum recommended human dose on a mg/m2 basis.
There are no adequate and well-controlled studies in pregnant women. PROCARDIA should be used during pregnancy only if the potential benefit justifies the potential risk.
Lactation
Nifedipine is transferred through breast milk. PROCARDIA should be used during breast-feeding only if the potential benefit justifies the potential risk.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established. Use in pediatric population is not recommended.
Geriatric Use
Age appears to have a significant effect on the pharmacokinetics of nifedipine. The clearance is decreased resulting in a higher AUC in the elderly. These changes are not due to changes in renal function (see CLINICAL PHARMACOLOGY, Pharmacokinetics).
ADVERSE REACTIONS
In multiple-dose United States and foreign controlled studies in which adverse reactions were reported spontaneously, adverse effects were frequent but generally not serious and rarely required discontinuation of therapy or dosage adjustment. Most were expected consequences of the vasodilator effects of PROCARDIA.
| PROCARDIA (%) | Placebo (%) | |
|---|---|---|
| Adverse Effect | (N=226) | (N=235) |
|
Dizziness, lightheadedness, giddiness |
27 |
15 |
|
Flushing, heat sensation |
25 |
8 |
|
Headache |
23 |
20 |
|
Weakness |
12 |
10 |
|
Nausea, heartburn |
11 |
8 |
|
Muscle cramps, tremor |
8 |
3 |
|
Peripheral edema |
7 |
1 |
|
Nervousness, mood changes |
7 |
4 |
|
Palpitation |
7 |
5 |
|
Dyspnea, cough, wheezing |
6 |
3 |
|
Nasal congestion, sore throat |
6 |
8 |
There is also a large uncontrolled experience in over 2100 patients in the United States. Most of the patients had vasospastic or resistant angina pectoris, and about half had concomitant treatment with beta-adrenergic blocking agents. The most common adverse events were:
Incidence Approximately 10%
Cardiovascular: peripheral edema
Central Nervous System: dizziness or lightheadedness
Gastrointestinal: nausea
Systemic: headache and flushing, weakness
Incidence Approximately 5%
Cardiovascular: transient hypotension
Incidence 2% or Less
Cardiovascular: palpitation
Respiratory: nasal and chest congestion, shortness of breath
Gastrointestinal: diarrhea, constipation, cramps, flatulence
Musculoskeletal: inflammation, joint stiffness, muscle cramps
Central Nervous System: shakiness, nervousness, jitteriness, sleep disturbances, blurred vision, difficulties in balance
Other: dermatitis, pruritus, urticaria, fever, sweating, chills, sexual difficulties
Incidence Approximately 0.5%
Cardiovascular: syncope (mostly with initial dosing and/or an increase in dose), erythromelalgia
Incidence Less Than 0.5%
Hematologic: thrombocytopenia, anemia, leukopenia, purpura
Gastrointestinal: allergic hepatitis
Face and Throat: angioedema (mostly oropharyngeal edema with breathing difficulty in a few patients), gingival hyperplasia
CNS: depression, paranoid syndrome
Special Senses: transient blindness at the peak of plasma level, tinnitus
Urogenital: nocturia, polyuria
Other: arthritis with ANA (+), exfoliative dermatitis, gynecomastia
Musculoskeletal: myalgia
Several of these side effects appear to be dose related. Peripheral edema occurred in about one in 25 patients at doses less than 60 mg per day and in about one patient in eight at 120 mg per day or more. Transient hypotension, generally of mild to moderate severity and seldom requiring discontinuation of therapy, occurred in one of 50 patients at less than 60 mg per day and in one of 20 patients at 120 mg per day or more.
Very rarely, introduction of PROCARDIA therapy was associated with an increase in anginal pain, possibly due to associated hypotension. Transient unilateral loss of vision has also occurred.
In addition, more serious adverse events were observed, not readily distinguishable from the natural history of the disease in these patients. It remains possible, however, that some or many of these events were drug related. Myocardial infarction occurred in about 4% of patients and congestive heart failure or pulmonary edema in about 2%. Ventricular arrhythmias or conduction disturbances each occurred in fewer than 0.5% of patients.
In a subgroup of over 1000 patients receiving PROCARDIA with concomitant beta blocker therapy, the pattern and incidence of adverse experiences were not different from that of the entire group of PROCARDIA (nifedipine) treated patients. (See PRECAUTIONS.)
In a subgroup of approximately 250 patients with a diagnosis of congestive heart failure as well as angina pectoris (about 10% of the total patient population), dizziness or lightheadedness, peripheral edema, headache, or flushing each occurred in one in eight patients. Hypotension occurred in about one in 20 patients. Syncope occurred in approximately one patient in 250. Myocardial infarction or symptoms of congestive heart failure each occurred in about one patient in 15. Atrial or ventricular dysrhythmias each occurred in about one patient in 150.
In post-marketing experience, there have been rare reports of exfoliative dermatitis caused by nifedipine. There have been rare reports of exfoliative or bullous skin adverse events (such as erythema multiforme, Stevens-Johnson Syndrome, and toxic epidermal necrolysis) and photosensitivity reactions. Acute generalized exanthematous pustulosis also has been reported.
OVERDOSAGE
Experience with nifedipine overdosage is limited. Generally, overdosage with nifedipine leading to pronounced hypotension calls for active cardiovascular support including monitoring of cardiovascular and respiratory function, elevation of extremities, and judicious use of calcium infusion, pressor agents, and fluids. Clearance of nifedipine would be expected to be prolonged in patients with impaired liver function. Since nifedipine is highly protein bound, dialysis is not likely to be of any benefit; however, plasmapheresis may be beneficial.
DOSAGE AND ADMINISTRATION
The dosage of PROCARDIA needed to suppress angina and that can be tolerated by the patient must be established by titration. Excessive doses can result in hypotension.
Therapy should be initiated with the 10 mg capsule. The starting dose is one 10 mg capsule, swallowed whole, 3 times/day. The usual effective dose range is 10–20 mg three times daily. Some patients, especially those with evidence of coronary artery spasm, respond only to higher doses, more frequent administration, or both. In such patients, doses of 20–30 mg three or four times daily may be effective. Doses above 120 mg daily are rarely necessary. More than 180 mg per day is not recommended.
In most cases, PROCARDIA titration should proceed over a 7–14 day period so that the physician can assess the response to each dose level and monitor the blood pressure before proceeding to higher doses.
If symptoms so warrant, titration may proceed more rapidly provided that the patient is assessed frequently. Based on the patient's physical activity level, attack frequency, and sublingual nitroglycerin consumption, the dose of PROCARDIA may be increased from 10 mg t.i.d. to 20 mg t.i.d. and then to 30 mg t.i.d. over a three-day period.
In hospitalized patients under close observation, the dose may be increased in 10 mg increments over four- to six-hour periods as required to control pain and arrhythmias due to ischemia. A single dose should rarely exceed 30 mg.
Avoid co-administration of nifedipine with grapefruit juice (see CLINICAL PHARMACOLOGY and PRECAUTIONS: Other Interactions).
No "rebound effect" has been observed upon discontinuation of PROCARDIA. However, if discontinuation of PROCARDIA is necessary, sound clinical practice suggests that the dosage should be decreased gradually with close physician supervision.
Co-Administration with Other Antianginal Drugs
Sublingual nitroglycerin may be taken as required for the control of acute manifestations of angina, particularly during PROCARDIA titration. See PRECAUTIONS, Drug Interactions, for information on co-administration of PROCARDIA with beta blockers or long-acting nitrates.
HOW SUPPLIED
PROCARDIA soft gelatin capsules are supplied in:
Bottles of 100: 10 mg (NDC 0069‑2600‑66)
The capsules should be protected from light and moisture and stored at controlled room temperature, 59° to 77°F (15° to 25°C) in the manufacturer’s original container.
LAB-0178-8.0
Revised November 2023
PRINCIPAL DISPLAY PANEL - 10 mg Capsule Bottle Label
Pfizer
NDC 0069-2600-66
Procardia
®
nifedipine
10 mg
Capsules
100 Capsules
Rx only
